In early access Dec 2006 Nucl Acid Res, researchers from 9 laboratories in France, Italy, Norway, and Denmark examine a candidate barcode gene for land plants, the group I intron in the chloroplast leucine transfer RNA gene (trnL intron). Prior research has already shown that a simultaneous or tiered multi-gene approach will be needed to distinguish among closely-related land plant species. A project coordinated by Royal Botanic Gardens, Kew aims to identify the best overall approach.
 Rather than cracking the tough nut of an ideal plant barcode, Taberlet and co-authors look at a simple approach “emphasizing the point of view of scientists other than taxonomists“, and test this on food plants in archeological and industrial applications. The chloroplast trnL intron is not the most variable non-coding region in chloroplast DNA and does not differ enough to separate many closely-related plant species. On the plus side, there are robust primers which amplify the intron from diverse species. Like other group I introns, the trnL intron sequence has catalytic activity and a conserved secondary structure with alternating conserved and variable sequence domains. Taking advantage of this feature, the researchers designed primers to amplify one of the variable domains, the P6 loop. Binding sites for both the trnL primers, which amplify the entire intron, and the P6 loop primers are “highly conserved among land plants, from Angiosperms to Bryophytes“. Importantly, the P6 loop is only 10 to 143 bp and can be amplified from degraded DNA.
Rather than cracking the tough nut of an ideal plant barcode, Taberlet and co-authors look at a simple approach “emphasizing the point of view of scientists other than taxonomists“, and test this on food plants in archeological and industrial applications. The chloroplast trnL intron is not the most variable non-coding region in chloroplast DNA and does not differ enough to separate many closely-related plant species. On the plus side, there are robust primers which amplify the intron from diverse species. Like other group I introns, the trnL intron sequence has catalytic activity and a conserved secondary structure with alternating conserved and variable sequence domains. Taking advantage of this feature, the researchers designed primers to amplify one of the variable domains, the P6 loop. Binding sites for both the trnL primers, which amplify the entire intron, and the P6 loop primers are “highly conserved among land plants, from Angiosperms to Bryophytes“. Importantly, the P6 loop is only 10 to 143 bp and can be amplified from degraded DNA.
Using “simulated ePCR” with the large GenBank data set, trnL intron and P6 loop sequences identified to species level 67% and 19% of cases respectively. However, in many practical applications, the number of possible species that need to be distinguished is relatively small and they are taxonomically diverse. Following this reasoning, Taberlet et al tested the intron and its P6 loop on a set of 132 species found in the Arctic and 72 species representing the commonest food plants. With Arctic plants, trnL intron and P6 identified to species level 85% and 47%, respectively. With the food data set, the tiny P6 loop was sufficient to identify 78% to species level. The P6 loop was successfully amplified from a 20,000-year old permafrost sample, from human feces, and from various processed foods including detecting potato, leek, and onion DNA in dried soup mix!
This is an exciting study, and DNA barcoding will likely have multiple applications in food safety. Whether or not these exact gene regions are adopted, a standardized approach will enable widespread and inexpensive use.
 In
In 

 and what is needed to harness DNA taxonomy in general and DNA barcoding in particular to speed description of the estimated 80% of earth’s biodiversity that is at yet undescribed: “a feedback loop that [uses] discrepancies between DNA and other data to refine species descriptions..founded in existing theory of evolutionary biology and phylogenetics”
and what is needed to harness DNA taxonomy in general and DNA barcoding in particular to speed description of the estimated 80% of earth’s biodiversity that is at yet undescribed: “a feedback loop that [uses] discrepancies between DNA and other data to refine species descriptions..founded in existing theory of evolutionary biology and phylogenetics”
 Red seaweeds, kingdom Rhodophyta, are “weird, wonderful, and extremely ancient” organisms distantly related to plants (Tudge 2000 The Variety of Life). Multicellular red algae arose at least 1.2 billion years ago, predating the earliest multicellular animals by 600 million years. Visual identification is challenging, as “morphology can be highly variable within and between species, and conspicuous features with which they can be readily identified are often lacking. In addition, highly convergent morphology is commonly encountered. …Identification is further compounded by the complexities of red algal life histories, many of which have a heteromorphic alternation of generations. Different life history stages of species have frequently been described as separate species and have only been linked through observations of life histories in culture and use of molecular techniques” (
Red seaweeds, kingdom Rhodophyta, are “weird, wonderful, and extremely ancient” organisms distantly related to plants (Tudge 2000 The Variety of Life). Multicellular red algae arose at least 1.2 billion years ago, predating the earliest multicellular animals by 600 million years. Visual identification is challenging, as “morphology can be highly variable within and between species, and conspicuous features with which they can be readily identified are often lacking. In addition, highly convergent morphology is commonly encountered. …Identification is further compounded by the complexities of red algal life histories, many of which have a heteromorphic alternation of generations. Different life history stages of species have frequently been described as separate species and have only been linked through observations of life histories in culture and use of molecular techniques” (

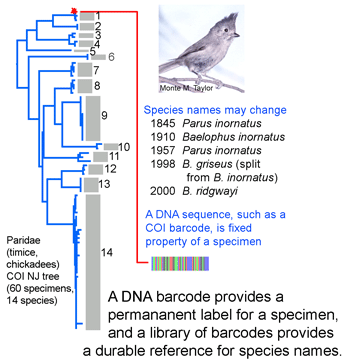 species names or boundaries, but that will not change DNA barcodes of specimens or the clustering patterns of barcode sequences. Thus it should be simple to use a specimen’s barcode sequence “name” to search a regularly revised public database for the current species name it corresponds to. A public database of sequences, specimens, and associated data as is
species names or boundaries, but that will not change DNA barcodes of specimens or the clustering patterns of barcode sequences. Thus it should be simple to use a specimen’s barcode sequence “name” to search a regularly revised public database for the current species name it corresponds to. A public database of sequences, specimens, and associated data as is