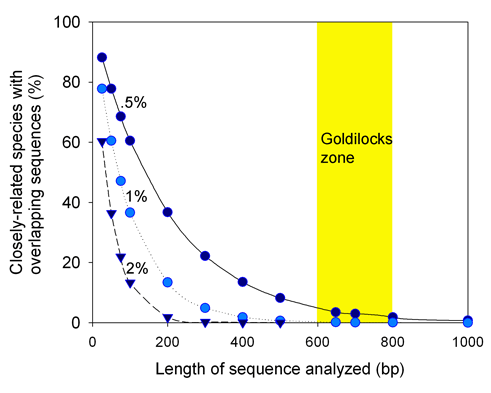
Collisions between birds and airplanes, known as birdstrikes , are an expensive hazard for civilian and military aircraft. Identification of airstrike specimens enables bird management near airfields and is essential for improvements in aircraft safety design. Forensic ornithology laboratories

(see for example, Laboratory for Feather Remains Identification in Tel Aviv) have relied on microscopic examination of feather barbules. Identification of birdstrikes through DNA barcoding seems likely to prove a reliable, reproducible, and rapid alternative. Here I try test flying a barcode approach, and compare to a Genbank BLAST search.
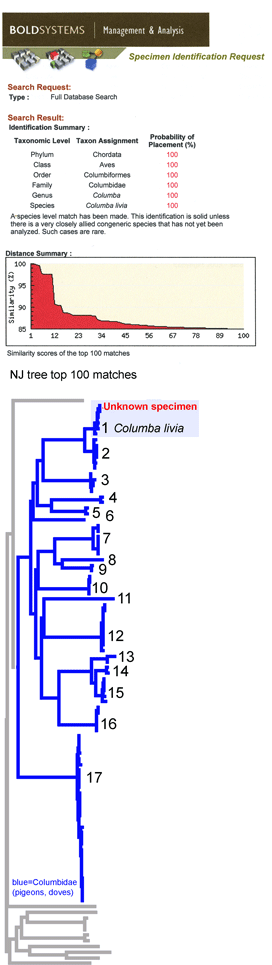 This simulation tries out what barcode identification might be like once reference libraries are established, and corresponds to “species identification” (vs species discovery) in last week’s post. A sequence was selected from Barcodes of Life Data Systems (BOLD) (130,000 COI barcode sequences from 19,000 species so far) and pasted into public “Identification Engine” on BOLD home page.
This simulation tries out what barcode identification might be like once reference libraries are established, and corresponds to “species identification” (vs species discovery) in last week’s post. A sequence was selected from Barcodes of Life Data Systems (BOLD) (130,000 COI barcode sequences from 19,000 species so far) and pasted into public “Identification Engine” on BOLD home page.
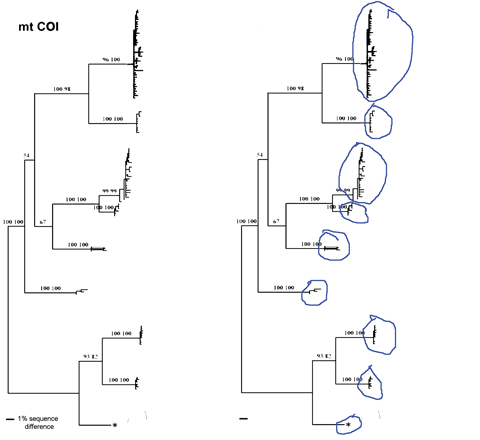
Voila! A probable identification with a disclaimer of infallibility, a list of the top 20 closest matches, and a graphic display of the closest 100 in the database. One more click creates a neighbor-joining tree with species names and collection sites (in the tree at left, species clusters are numbered, and the species and site names are omitted). 
Skipping over to Rock Pigeon Columba livia page at All Birds Barcoding Initiative (ABBI) website reveals a Google map of specimen locations.
So far the BOLD database contains sequences of 24 (8%) of the 309 Columbiformes (pigeons and doves) with an average of 4 specimens per species. More contributions will establish a comprehensive reference library.
A BLAST Genbank search with the C. livia COI sequence also shows C. livia as the closest match, but only a few closely-related birds. All COI sequences in BOLD are or will presumably be deposited in GenBank, but to date many are not yet public. For a more robust comparison, I tried a C. livia cytochrome b sequence, as cytb has historically been favored by vertebrate biologists (and COI by those studying invertebrates). The C. livia cytb sequence naturally matches most closely with C. livia, with C. rupestris as the sister species, the same pattern as with COI (in tree at left, C. rupestris is species 2). It is also possible to draw a NJ tree with results of BLAST search.
There are two obvious differences in the databases. First, Genbank BLAST output including the NJ tree does not show collection sites, which are helpful or essential when assessing variation within and among species. To find this information, one would have to go back to original publications which may be inacessible or not include this data, and many sequences are deposited without any published reference.
Second, in GenBank most species are represented by a single sequence. One of the strongest benefits of the barcode initiative, for those interested in population biology and species level-taxonomy, as well as for reliable identification, will be the collection of barcodes from multiple specimens for each species.