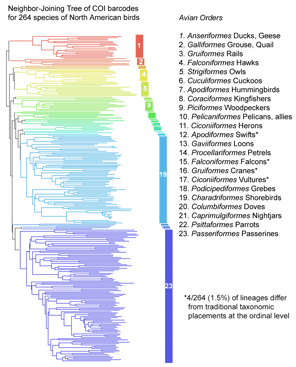
 COI barcoding is a standardized approach to identifying species by DNA, helping resolve the “leaves” on the tree of life. Will the growing arrays of COI sequences also help provide insight into evolutionary history, the “branches” of the tree? I am struck that in some cases, simple genetic arithmetic with COI sequences creates trees very similar to modern phylogenies painstakingly created from multiple nuclear and mitochondrial genes, multiple morphologic characters, and exhaustive computerized analysis. Shown at right, a neighbor-joining analysis of COI barcodes of 264 species of North American birds creates a tree that is quite similar to the most recent understanding of relationships among modern birds, with anseriformes (ducks and geese) next to galliformes (grouse and quail) at the top, passeriformes (perching birds) at the bottom, and most of the other established orders appearing as single lineages in between. Most of the families and groupings of families within these orders also match current understanding, including for example, that flycatchers appear as the basal lineage within passeriformes, and a group of New World passerines called nine-primaried oscines appear together at the bottom of the tree. Of course, a systematist would immediately note that the statistical support for these higher-order branches is weak or absent. I should tread lightly here or not at all, since phylogenetics is the province of mathematical experts, but I will plunge ahead anyway and suggest that, if the single gene neighbor-joining distance tree is “right” say 8 times out of 10, then the apparent lack of statistical support for higher order branches is misleading. There are cases where the COI gene tree is incorrect (eg Hajibabaei et al Genome 49:851 2006).
COI barcoding is a standardized approach to identifying species by DNA, helping resolve the “leaves” on the tree of life. Will the growing arrays of COI sequences also help provide insight into evolutionary history, the “branches” of the tree? I am struck that in some cases, simple genetic arithmetic with COI sequences creates trees very similar to modern phylogenies painstakingly created from multiple nuclear and mitochondrial genes, multiple morphologic characters, and exhaustive computerized analysis. Shown at right, a neighbor-joining analysis of COI barcodes of 264 species of North American birds creates a tree that is quite similar to the most recent understanding of relationships among modern birds, with anseriformes (ducks and geese) next to galliformes (grouse and quail) at the top, passeriformes (perching birds) at the bottom, and most of the other established orders appearing as single lineages in between. Most of the families and groupings of families within these orders also match current understanding, including for example, that flycatchers appear as the basal lineage within passeriformes, and a group of New World passerines called nine-primaried oscines appear together at the bottom of the tree. Of course, a systematist would immediately note that the statistical support for these higher-order branches is weak or absent. I should tread lightly here or not at all, since phylogenetics is the province of mathematical experts, but I will plunge ahead anyway and suggest that, if the single gene neighbor-joining distance tree is “right” say 8 times out of 10, then the apparent lack of statistical support for higher order branches is misleading. There are cases where the COI gene tree is incorrect (eg Hajibabaei et al Genome 49:851 2006).
I close with a picture inspired by the data. If single gene trees usually correspond to evolutionary history, this implies strong barriers to gene flow arise concurrent with differences in the single gene and are continuously maintained.




 A dozen articles in current issue of
A dozen articles in current issue of