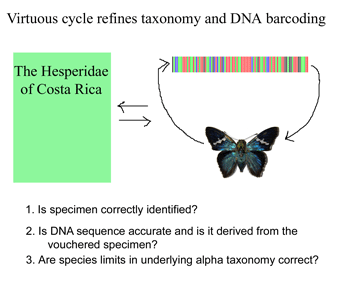
 Like a map that is regularly updated, the reliability of DNA barcode databases will improve over time. To enable improvement, researchers have agreed to standardize on a particular region, to analyze multiple individuals from each species, and to revise DNA sequences and taxonomic labels as new information becomes available. By using specimens archived in museums, taxonomic identifications and DNA sequences can be re-checked. In March 2007 Med Vet Entomol 21:44, researchers from University of Wollongong, Australia, apply DNA barcoding to the identification of 9 species of forensically and medically important blowflies in family Calliphoridae. Calliphoridae blowflires cause disease in humans and domestic animals, and, in cases of murder or suspicious death, identification of blowfly species is a first step in determining the post-mortem interval. Identifications of adult flies requires specialized taxonomic knowledge and even experts have difficulty identifying egg and larval stages and the fragments of decomposed insects that may be all that is available in forensics. Nelson et al sequenced COI barcode region from legs of 52 adult flies representing 9 species in genus Chrysoma. The specimens were deposited in the Diptera collection at the School of Biological Sciences, University of Wollongong. NJ and Bayesisan analyses recovered each species as a distinct cluster, ie a well-supported reciprocally monophyletic group.
Like a map that is regularly updated, the reliability of DNA barcode databases will improve over time. To enable improvement, researchers have agreed to standardize on a particular region, to analyze multiple individuals from each species, and to revise DNA sequences and taxonomic labels as new information becomes available. By using specimens archived in museums, taxonomic identifications and DNA sequences can be re-checked. In March 2007 Med Vet Entomol 21:44, researchers from University of Wollongong, Australia, apply DNA barcoding to the identification of 9 species of forensically and medically important blowflies in family Calliphoridae. Calliphoridae blowflires cause disease in humans and domestic animals, and, in cases of murder or suspicious death, identification of blowfly species is a first step in determining the post-mortem interval. Identifications of adult flies requires specialized taxonomic knowledge and even experts have difficulty identifying egg and larval stages and the fragments of decomposed insects that may be all that is available in forensics. Nelson et al sequenced COI barcode region from legs of 52 adult flies representing 9 species in genus Chrysoma. The specimens were deposited in the Diptera collection at the School of Biological Sciences, University of Wollongong. NJ and Bayesisan analyses recovered each species as a distinct cluster, ie a well-supported reciprocally monophyletic group.
Early in the study, two complications were encountered. First, four specimens preliminarily identified as Ch. latifrons grouped with Ch. semimetallica. Second, a specimen identified as Ch. saffranea grouped with its closest relative Ch. megacephala. The adult voucher specimens were re-examined and the nuclear ITS gene was sequenced for these individuals. This confirmed that the first four specimens had been misidentified, in retrospect unsurprising given the close morphological similarity of the two species. The fifth anomalous individual was diagnosed as a hybrid based on comparison of nuclear and mitochondrial sequences. The authors conclude “the need for re-examination of misplaced specimens…highlights the importance of a voucher collection for all members of a barcode database.” I would add that the researchers’ willingness to re-examine taxonomic identifications and sequence data is just as important as the availability of voucher specimens.
Two other recent papers on blowfly identification with mitochondrial DNA showed incomplete resolution at species level, but in these the authors did not close the taxonomy-DNA circle, either by re-examining specimens or repeating sequence analysis. In Int J Legal Med 2007, Wells et al examined Lucilia sp blowflies, using published GenBank sequences and newly sequenced adult flies, and found overlap between all sister species of Lucilia for which 2 or more specimens were examined. It is unclear from this short note how many specimens were examined, their geographic origin, and whether they are stored as vouchers (online supplementary material is not available on publisher’s website at the time of this writing). There is no mention of re-examining their own specimens or analyzing other loci and of course it is not possible in most cases to confirm that taxonomic identifications and sequences in GenBank data are correct.
In July 2007 Proc R Soc B Whitworth et al examine 31 Protocalliphora individuals belonging to 12 species. Protocalliphora are Holoarctic species whose larva parasitize newly-hatched nestling birds. Blowfly larvae or pupae were collected from nests, and emergent flies were identified based on fly and pupal case morphology. As “the lower half of the abdomen of each fly was used for DNA sequencing” I assume this would not leave enough tissue for voucher specimens. They first attempted to construct a phylogeny using nuclear ITS, but found a very low level of substitutions between species and those found were all autoapomorphies, both of which suggest this is a very recently derived species complex. They were able to construct a phylogeny using amplified fragment length polymorphism (AFLP) mapping, with each species forming a reciprocally monophyletic group. This was then compared to mitochondrial sequence data.
Given that the title of the paper is “DNA barcoding cannot reliably identify….” it is inexplicable and also scientically inaccurate that they did NOT analyze the standard 648 bp COI barcode region, instead using a 374 bp fragment of COI and a 579 fragment of COII! It is likely that the results would be similar in any case, but their mitochondrial data cannot be combined with or directly compared to results with the growing DNA barcode libraries, which now contain about 260,000 barcode records from about 29,000 species. The mitochondrial sequences showed distinct clusters for 6 of the 12 species, and there were 2 other clusters comprising 2 and 4 species respectively. A separate analysis suggests these multi-species clusters reflect horizontal transfer of mitochondrial DNA among closely-related species as a result of Wolbachia infection, and the authors speculate that, since Wolbachia are found in “15-75% of insect species”, there may difficulty using DNA barcoding to resolve many insect species. To my reading, their data suggest this is a very recently derived species complex and hybridization among species is common. One of the utilities of DNA barcoding is to highlight exceptional groups, such as this one appears to be, deserving of further study. For the next studies on DNA barcoding in Lucilia and Protocalliphora, I hope the researchers retain voucher specimens and sequence the standard barcode fragment!
 Growing data sets demonstrate DNA barcoding usually works, but why? Why does a very short stretch of DNA, such as a DNA barcode which usually represents less than one one-millionth of the genome, enable identification of most animal species? In computer language, Rod Page describes a DNA barcode as “
Growing data sets demonstrate DNA barcoding usually works, but why? Why does a very short stretch of DNA, such as a DNA barcode which usually represents less than one one-millionth of the genome, enable identification of most animal species? In computer language, Rod Page describes a DNA barcode as “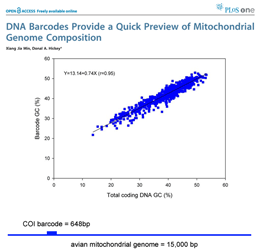 the DNA barcodes can provide a quick preview of the whole genome.” It will be of great interest to extend this analysis to compare mitochondrial barcodes to nuclear genomes; the general success of barcoding approach suggests there will be similarly close correlation.
the DNA barcodes can provide a quick preview of the whole genome.” It will be of great interest to extend this analysis to compare mitochondrial barcodes to nuclear genomes; the general success of barcoding approach suggests there will be similarly close correlation.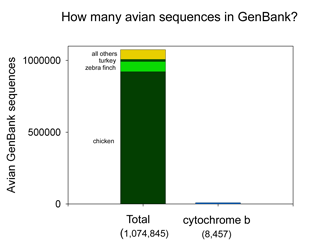 There are more than 1 million sequences in GenBank, but over 900,000 are from the Jungle Fowl (ie chicken, Gallus gallus), and another 85,000 from Zebra finch (Taeniopyga guttata) and Wild turkey (Meleagris gallopavo). That leaves about 67,000 sequences in total representing the rest of the approximately 10,000 species of world birds. According to Clements’ Birds of the World (including updates through 2006), there are 9,919 recognized species. The other world lists are very similar, and differ primarily in whether certain forms are recognized as species or subspecies and in assignment of generic names. I find it surprising there is not a single global taxonomic authority for bird species status, names, spelling, generic and family classification. As a comparison, medicine would be in great difficulty if there were not a single standard nomenclature for pathogenic bacteria.
There are more than 1 million sequences in GenBank, but over 900,000 are from the Jungle Fowl (ie chicken, Gallus gallus), and another 85,000 from Zebra finch (Taeniopyga guttata) and Wild turkey (Meleagris gallopavo). That leaves about 67,000 sequences in total representing the rest of the approximately 10,000 species of world birds. According to Clements’ Birds of the World (including updates through 2006), there are 9,919 recognized species. The other world lists are very similar, and differ primarily in whether certain forms are recognized as species or subspecies and in assignment of generic names. I find it surprising there is not a single global taxonomic authority for bird species status, names, spelling, generic and family classification. As a comparison, medicine would be in great difficulty if there were not a single standard nomenclature for pathogenic bacteria. 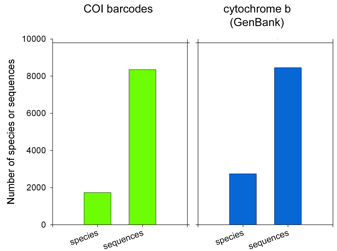 62,571 of the remaining 66,969 sequences are in the “CoreNucleotide” database (the others are unnamed genetic loci, either Expressed Sequence Tag (EST) or Genome Survey Sequence (GSS) records, and these will not be considered further here). Only 4,951 bird species are represented by any sequence (50% of world birds), and there are cytochrome b sequences for only 2,751 species (28% of world birds). Of species with cyt b sequences, 60% are represented by single sequences.
62,571 of the remaining 66,969 sequences are in the “CoreNucleotide” database (the others are unnamed genetic loci, either Expressed Sequence Tag (EST) or Genome Survey Sequence (GSS) records, and these will not be considered further here). Only 4,951 bird species are represented by any sequence (50% of world birds), and there are cytochrome b sequences for only 2,751 species (28% of world birds). Of species with cyt b sequences, 60% are represented by single sequences.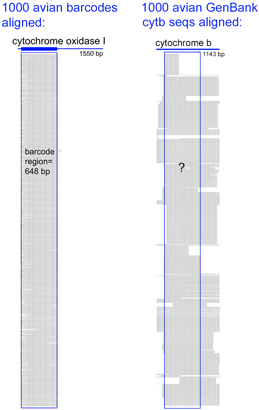 Virtues of the DNA barcode data set include that sequences are linked to vouchered museum specimens and their associated collecting data, sequence records include trace files to confirm sequencing accuracy, and most important all sequences can be directly compared because they derive from a standardized region. GenBank cyt b files include sequences of varying length and position along the gene. An alignment of 1000 avian COI barcodes and 1000 avian cyt b sequences hints at the power of a standardized approach.
Virtues of the DNA barcode data set include that sequences are linked to vouchered museum specimens and their associated collecting data, sequence records include trace files to confirm sequencing accuracy, and most important all sequences can be directly compared because they derive from a standardized region. GenBank cyt b files include sequences of varying length and position along the gene. An alignment of 1000 avian COI barcodes and 1000 avian cyt b sequences hints at the power of a standardized approach. A dream of many came to life this week with launch of
A dream of many came to life this week with launch of  The Scanning and Digitization Group will accelerate the work of the
The Scanning and Digitization Group will accelerate the work of the 
 In
In 
 The results show a “variegated picture of the taxonomic status of publicly indexed fungal sequences“. Taxonomic coverage is sparse: of the estimated 1.5 million fungi, less than 1% (9,684 species) are represented. Taxonomic data is lacking for many sequences (27% are not identified to species level), and most of the species-level identifications are unverifiable (82% are not linked to voucher specimens, 63% are not tagged with specimen country of origin, and 42% are marked as unpublished). Sequence comparisions suggest mislabeling is common (11% show best matches to congeneric but heterospecific sequences, and another 7% match among species of a different genus. Overall 10-21% of the INSD sequences have incorrect or unsatisfactory annotations.
The results show a “variegated picture of the taxonomic status of publicly indexed fungal sequences“. Taxonomic coverage is sparse: of the estimated 1.5 million fungi, less than 1% (9,684 species) are represented. Taxonomic data is lacking for many sequences (27% are not identified to species level), and most of the species-level identifications are unverifiable (82% are not linked to voucher specimens, 63% are not tagged with specimen country of origin, and 42% are marked as unpublished). Sequence comparisions suggest mislabeling is common (11% show best matches to congeneric but heterospecific sequences, and another 7% match among species of a different genus. Overall 10-21% of the INSD sequences have incorrect or unsatisfactory annotations.