Goldilocks finds mtDNA COI barcode length “just right” for distinguishing most animal species, asks why
Friday, August 25th, 2006
The standard animal barcode 648 bp of mitochondrial gene COI seems “just right” for delimiting most animal species. If it were “too short”, then closely-related species would not be resolved. If it were “too long” then sequencing effort would be wasted. Here I examine what might underlie the Goldilocks effect.
The following figure looks at how often closely-related species (differing by .5%, 1%, or 2%) are predicted to have overlapping sequences. With the assumptions examined below, above 600 base pairs all but the most-closely-related species will be distinguished, and above 800 base pairs, there is little gain in sensitivity.
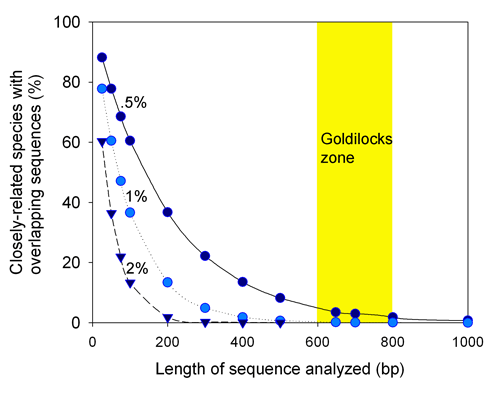
The assumptions underlying this table-napkin analysis appear supported by data so far:
First, mitochondrial DNA sequence differences between closely-related species are widely and relatively evenly distributed throughout the protein coding and ribosomal genes. For example, see an earlier post with percent identity plots comparing whole mitochondrial genomes for congeneric salamanders. Further support is provided by a plot of parallel sequence differences in the 2 most commonly utilized mitochondrial genes, COI and cytB.
Second, most closely-related animal species have COI sequences that differ by at least 1%. For example more than 98% of 13,320 congeneric pairs from a wide array of invertebrate and vertebrate species showed greater than 2% sequence difference (Hebert et al 2003 Proc Biol Sci 270:S96).
Third, intraspecific sequence variation in mtDNA is generally very low, less than 1% in most animal species.
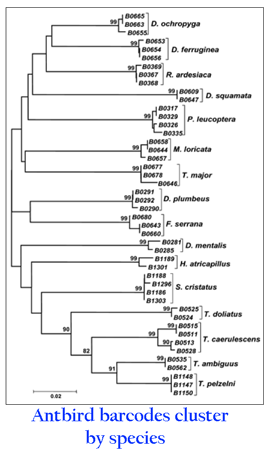
If most closely-related species can be distinguished by short mtDNA sequences, then recognizing the sets of mtDNA sequences that make up species, ie species delimitation, should at least sometimes be simple. Using the neighbor joining tree of mtDNA barcodes below, an untrained person might pick out the groups of sequences that correspond to species. The top 5 groups represent previously unrecognized cryptic species of scorched mussel Brachidontes exustus (Lee and Foighil 2004 Mol Ecol 13:3527)
Goldilocks leaves us with the scientific questions: why are differences within most species so small, and why are the distances between most nearest neighbor species so large?







 Do you see a new species anywhere? Baleen whale specimen collected in 1976, new species description 27 years later based in part on mitochondrial DNA characters (Wada et al. 2003. Nature 426:278)
Do you see a new species anywhere? Baleen whale specimen collected in 1976, new species description 27 years later based in part on mitochondrial DNA characters (Wada et al. 2003. Nature 426:278) 
 With funding from the Gordon and Betty Moore Foundation, researchers at Reveo, Inc. and the University of Washington are collaborating on developing a
With funding from the Gordon and Betty Moore Foundation, researchers at Reveo, Inc. and the University of Washington are collaborating on developing a