In July 2 PLOS ONE article, “DNA barcoding works in practice but not in (neutral) theory,” David Thaler and I argue a radically different view of how evolution works, as compared to the standard neutral model, is needed to account for the widespread pattern of limited variation within species and larger differences among that underlies the general effectiveness of DNA barcoding. The following text is adapted from the article.
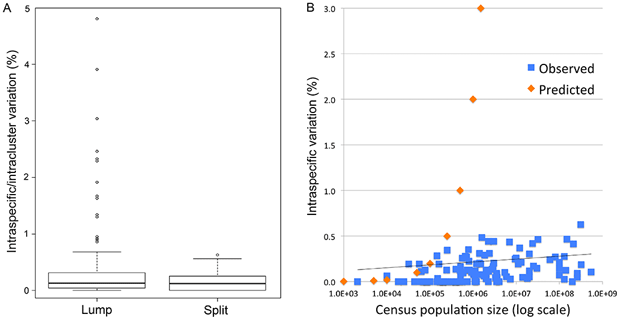
Fig. 1 (from PLOS ONE article). Intraspecific variation in birds is uniformly low across 100,000-fold differences in census population size. Apparent outliers reflect lumping of reproductively isolated populations.
“To to better understand the limits to DNA barcoding and the evolutionary mechanisms that underlie the usual barcode gap pattern, we used birds to test whether differences within and among species conform to neutral theory, the reigning null hypothesis for mitochondrial sequence evolution. We analyzed apparent barcode gap exceptions in detail–those with unusually large intraspecific differences and those lacking interspecific differences.
From a practical point of view exceptions may help define limits to COI barcodes as a marker of speciation. In the context of evolutionary theory, exceptions may give valuable insight into the mechanisms controlling variance within and among species. Birds are uniquely suited this task: they are well represented in barcode libraries, have the best-known species limits of any large animal group, and, most critically, are the only large group with known census population sizes, a key parameter in neutral theory.
Neutral theory predicts intraspecific variation equals 2 Nµ, where N is population size and µ is mutation rate per generation. Although textbooks and scientific reports recognize a multitude of exceptions to this predicted relationship, deviations are subsumed under the rubric of “effective population size” and accounted for by ad hoc modifications to the theory, which is assumed operative.
Here we harness the unique resources of avian barcode libraries and census population data to look at the question the other way around, namely, do the empirical data show any signature of variance proportional to population size? If not, does the observed range of variation fit with commonly proposed modifications to neutral theory? In addition, we examine whether molecular clock measurements conform to neutral theory prediction that clock rate equals µ.
This is the first large study of animal mitochondrial diversity using actual census population sizes and the first to test outliers for population structure. We demonstrate uniformly low intraspecific mitochondrial DNA variation in birds regardless of population size. Nearly all apparent exceptions reflect lumping of reproductively isolated populations (many of which represent distinct species) or hybrid lineages. To our knowledge, this is the first large test of neutral theory applied to mitochondrial diversity using actual census population measurements rather than crude proxies of population size such as phylogeny or body weight, and the first to test outliers for population structure.
In contrast to prior analyses, we find uniformly low intraspecific variation regardless of census population size. Universally low intraspecific variation contradicts a central prediction of neutral theory and is not readily accounted for by commonly proposed ad hoc modifications. We conclude that this finding together with the molecular clock phenomenon are strong evidence that neutral processes play a minor role in animal mitochondrial evolution.
We argue a radically different view of evolution–extreme purifying selection and continuous adaptive evolution–is needed to account for the widespread pattern of limited variation within species and larger differences among that underlies the general effectiveness of DNA barcoding.”
I hope you enjoy!










{kind=link}
{kind=link}