The fastest way forward
Thursday, November 2nd, 2006
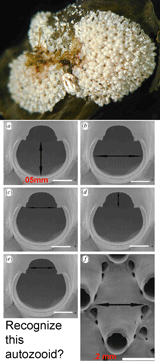 In October Proc R Soc B Gomez et al apply DNA barcoding to the cosmopolitan marine bryozoan Celleporella hyalina. Morphologic identification in this genus uses scanning electron microscopy measurements of the 0.2 mm autozooid and its 0.05 mm orifice. To eliminate potential variability associated with colonial development or environmental plasticity, these morphologic measurements are made on cloned F1 progeny grown under controlled laboratory conditions. This example highlights how standard morphologic techniques can be cumbersome and costly, and require highly-trained personnel and expensive equipment. It is unlikely this sort of morphologic identification process can be sped up, while DNA analysis is getting faster, cheaper, and more portable.
In October Proc R Soc B Gomez et al apply DNA barcoding to the cosmopolitan marine bryozoan Celleporella hyalina. Morphologic identification in this genus uses scanning electron microscopy measurements of the 0.2 mm autozooid and its 0.05 mm orifice. To eliminate potential variability associated with colonial development or environmental plasticity, these morphologic measurements are made on cloned F1 progeny grown under controlled laboratory conditions. This example highlights how standard morphologic techniques can be cumbersome and costly, and require highly-trained personnel and expensive equipment. It is unlikely this sort of morphologic identification process can be sped up, while DNA analysis is getting faster, cheaper, and more portable.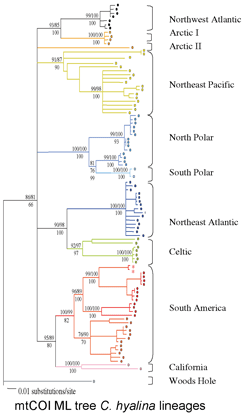
The researchers from University of Hull, University of Wales, and Universidad Catolica de la Santisima Concepcion in Chile analyzed mtCOI barcodes in 176 colonies from 33 sites around the globe, revealing at least 10 deeply divergent lineages. Mating compatability in 26 pairwise combinations showed complete reproductive isolation in 23 cases, and 3 were inconclusive due to self-fertilization. Only one of the genetically divergent, reproductively incompatible groups could be reliably separated by morphologic analysis.
It is obviously impractical to do mating studies for routine identification of bryozoans. Instead, standardized genetic analysis, ie DNA barcoding, can first help discover species (as in this case by highlighting lineages that were then subjected to other forms of biological analysis), and then be applied to assign unknown specimens to the newly revealed species. The authors conclude “DNA barcoding clearly identifies biologically meaningful groups in the C. hyalina complex” and speculate that biodiversity is similarly underestimated in other sessile marine invertebrates, including sponges and corals. “Failure to recongize cryptic speciation among sessile benthos therefore may seriously underestimate marine biodiversity as well as impeding attempts to predict the response of marine benthos to environmental change.” I conclude that DNA barcoding is the fastest way forward to help discover and then routinely identify what appear to be the vast numbers of cryptic animal species.
 The Sponge Barcoding Project
The Sponge Barcoding Project 
 In 1987, the few dozen GPS models available were mostly larger than 200 cu in and cost $15,000 to $45,000. (
In 1987, the few dozen GPS models available were mostly larger than 200 cu in and cost $15,000 to $45,000. ( Good news for taxonomic science:
Good news for taxonomic science: 
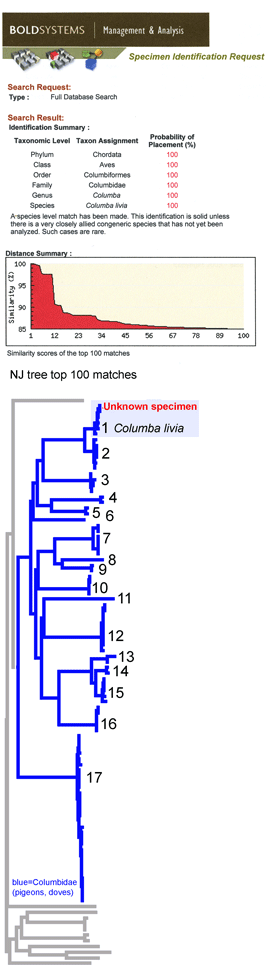 This simulation tries out what barcode identification might be like once reference libraries are established, and corresponds to “species identification” (vs species discovery) in last week’s post. A sequence was selected from Barcodes of Life Data Systems (BOLD) (130,000 COI barcode sequences from 19,000 species so far) and pasted into public “Identification Engine” on
This simulation tries out what barcode identification might be like once reference libraries are established, and corresponds to “species identification” (vs species discovery) in last week’s post. A sequence was selected from Barcodes of Life Data Systems (BOLD) (130,000 COI barcode sequences from 19,000 species so far) and pasted into public “Identification Engine” on