Two recent articles suggest how and how not to learn about applying mtCOI sequences to identifying species. In Zoologica Scripta 2006 35:441 researchers from Koenig Zoological Research Museum, Bonn, analyze 113 specimens of 61 morphologically-defined species of pholcid (daddy long-legs) spiders. Important for this analysis and for future study, collection locations are given and voucher numbers are provided for each specimen and DNA extract.
Two recent articles suggest how and how not to learn about applying mtCOI sequences to identifying species. In Zoologica Scripta 2006 35:441 researchers from Koenig Zoological Research Museum, Bonn, analyze 113 specimens of 61 morphologically-defined species of pholcid (daddy long-legs) spiders. Important for this analysis and for future study, collection locations are given and voucher numbers are provided for each specimen and DNA extract.
(Some pholcid spiders vibrate in their webs when disturbed, moving so rapidly they become invisible; here is a wonderful video)
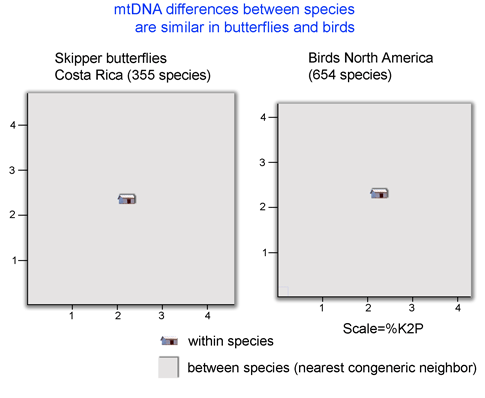
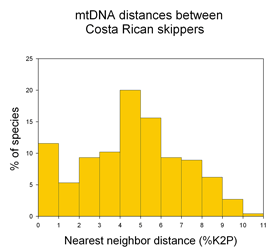
16s and COI sequences were successfully amplified using a single primer pair for each gene from 79% and 80% of specimens, respectively. It is striking that strong clustering within species was observed despite using short segments of mtDNA (COI, 312 bp; 16s 287 bp), which are less than half as long as the standard 648 bp COI barcode. In NJ trees with either mtDNA sequence, all morphologic conspecifics grouped together and were reciprocally monophyletic (ie no overlaps between species). Likely splits based on large intraspecific distances and differing geographic distributions were observed in 6 (25%) of 24 multiply-sampled morphospecies.
The authors go on to propose graphic and statistical metrics to calibrate how well simple distances can define species limits. They find that mtDNA distances will often diagnose species: “tree-based taxon clustering and statistical taxon analysis indicate that molecular evidence does coincide with morphological hypotheses” and “we disagree with [Meyer and Paulay’s] point that independently of the group of organisms studied, a “barcoding gap” between interspecific and intraspecific distance values would likely disappear in studies featuring both dense within-species sampling and closely related species”, ie distance-based clustering often corresponds to species limits.
This study uses vouchered specimens from known locations and accurate modern sequencing technology, focuses on a relatively small clade (959 known species pholcid spiders), and analyzes in a positive way how distance measures might be used to define species, helping us learn about DNA barcoding as a tool for species identification.
In another recent study Syst Biol 55:715 2006 researchers from National University of Singapore examine COI sequences deposited in GenBank from Diptera (flies, mosquitos, and gnats). They found 449 of the 150,000 known species of dipterans represented, with multiple sequences from 127 species, and analyze these to “test two key claims of molecular taxonomy”. The scientists found that there were often large differences in COI within species and also frequent overlaps between species such that some sequences were more closely related to those of another species than to conspecifics. The litany of failures is quite long, including “even when two COI sequences are identical, there is a 6% chance they belong to different species”.
I do not understand why the authors put so much effort into analyzing such a heterogeneous set of data, except that they are worried about molecular taxonomy in general and DNA barcoding in particular. To my reading this study suggests that many GenBank records contain errors, either because current morphologic taxonomy is incorrect (for example, study cited above suggests probable splits in 25% of pholcid spiders), specimens used for GenBank records are incorrectly identified, or because DNA sequences in GenBank contain errors due to human factors or older sequencing technology. There must be some limitations to COI barcode identification of dipteran species, mostly presumably closely-related young species, but this study has not shown where such problems might lie.
I hope that future studies will use more of the “best practices” demonstrated in Astrin et al’s study of pholcid spiders and so help us learn more about how to apply COI sequences to species identification.


 Insiders can be mistaken, in science and in other fields. At the beginning of the Human Genome Project, “the great majority of scientists dismissed the original proposal with hostility or indifference” (
Insiders can be mistaken, in science and in other fields. At the beginning of the Human Genome Project, “the great majority of scientists dismissed the original proposal with hostility or indifference” (
 The Sponge Barcoding Project
The Sponge Barcoding Project 
 In 1987, the few dozen GPS models available were mostly larger than 200 cu in and cost $15,000 to $45,000. (
In 1987, the few dozen GPS models available were mostly larger than 200 cu in and cost $15,000 to $45,000. ( Good news for taxonomic science:
Good news for taxonomic science: 
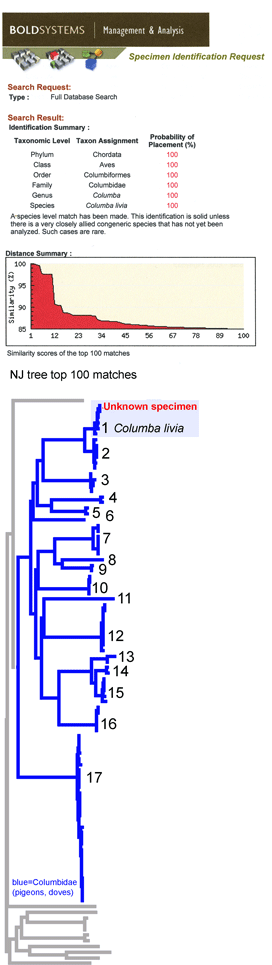 This simulation tries out what barcode identification might be like once reference libraries are established, and corresponds to “species identification” (vs species discovery) in last week’s post. A sequence was selected from Barcodes of Life Data Systems (BOLD) (130,000 COI barcode sequences from 19,000 species so far) and pasted into public “Identification Engine” on
This simulation tries out what barcode identification might be like once reference libraries are established, and corresponds to “species identification” (vs species discovery) in last week’s post. A sequence was selected from Barcodes of Life Data Systems (BOLD) (130,000 COI barcode sequences from 19,000 species so far) and pasted into public “Identification Engine” on