The Technology-Entertainment-Design (TED) conference that helped launch the Encyclopedia of Life and connect it with hi-techsters prepared a 4-minute, 13 MB video [download it here] that both reports on progress and shows some nifty features in store for future EOL users.
News
Routine DNA ID as quality control in ecology and evolutionary biology
 Just as DNA analysis regularly overturns seemingly solid eyewitness identifications in crime investigations, routine DNA analysis can also help biologists avoid blunders. In 28 August 2007 Mol Ecol, researchers from University of Colorado, New Mexico State University, Pisces Molecular, and Brigham Young University report that over 20 years of restocking efforts in western US aimed at restoring native populations of endangered greenback cutthroat trout Oncorhynchus clarkii stomias have mostly been restocking a non-native, non-endangered subspecies, Colorado River cutthroat trout O. c. pleuriticus. They trace the confusion to repeated introductions beginning in the late 1800s of Colorado River cutthroat trout throughout the native range of greenback cutthroat trout. The authors analyzed mitochondrial (COI, ND2) and nuclear (microsatellites, AFLP) DNA from 365 individuals from 15 locations in 3 major river drainage systems in Colorado and surrounding states. Distinct mtDNA lineages corresponding to each subspecies were corroborated by nuclear microsatellite and AFLP data. For another cautionary tale of repeated misidentification of a widely studied organism, see Siddall and colleagues’ entertaining June 2007 Proc R Soc paper scrutinizing commercially available medicinal leeches sold as Hirudo medicinalis.
Just as DNA analysis regularly overturns seemingly solid eyewitness identifications in crime investigations, routine DNA analysis can also help biologists avoid blunders. In 28 August 2007 Mol Ecol, researchers from University of Colorado, New Mexico State University, Pisces Molecular, and Brigham Young University report that over 20 years of restocking efforts in western US aimed at restoring native populations of endangered greenback cutthroat trout Oncorhynchus clarkii stomias have mostly been restocking a non-native, non-endangered subspecies, Colorado River cutthroat trout O. c. pleuriticus. They trace the confusion to repeated introductions beginning in the late 1800s of Colorado River cutthroat trout throughout the native range of greenback cutthroat trout. The authors analyzed mitochondrial (COI, ND2) and nuclear (microsatellites, AFLP) DNA from 365 individuals from 15 locations in 3 major river drainage systems in Colorado and surrounding states. Distinct mtDNA lineages corresponding to each subspecies were corroborated by nuclear microsatellite and AFLP data. For another cautionary tale of repeated misidentification of a widely studied organism, see Siddall and colleagues’ entertaining June 2007 Proc R Soc paper scrutinizing commercially available medicinal leeches sold as Hirudo medicinalis.
How might the future look with routine application of DNA ID as quality control? Incorporating DNA barcode analysis into Tree of Life studies is one useful approach, exemplified by two recent large-scale evolutionary studies published in January and April 2008 Syst Entomol, one on phylogenetic relationships in Saturnid silkmoths, and one on higher-level relationships among 12 families in ‘bombycoid complex’ of Lepidoptera. Both studies analyze COI barcodes of all specimens, “allowing confirmination of their identification for species present in the BOLD reference library and enabling future identifications of organisms whose identity is still pending.”
Interview by Heinz Horeis
German journalist Heinz Horeis who specializes in energy and environment visited the PHE in late 2007. The Swiss weekly news magazine Weltwoche published in German 6 March 2008 a substantial version of Heinz’s longer English conversation with Jesse. A couple of excerpts:
“In twenty years, [renewable] sources will have failed economically, leaving renewable energy to be remembered as the energy equivalent of sub-prime mortgages. “
“But humans are not rational. Why do people buy lottery tickets? They hope for a solution, effectively by magic, as lottery jackpot odds are one in millions. Much of the enthusiasm for renewables is belief in magic. People tire of hearing about problems related to fossil fuels or nuclear power, presented in great detail for 30 years. Anything different sounds better. Humans want to believe. In a profound short story called the Kugelmass Episode by the American humorist Woody Allen, the dissatisfied hero rejects the psychoanalyst who has been trying to adjust Kugelmass to reality and chooses to patronize a magician instead, who works the miracle of transporting Kugelmass into the novel Madame Bovary, with whom Kugelmass then has an affair. Kugelmass, biomass.”
Photo from Encyclopedia of Life launch
Photo from With the Encyclopedia of Life launched, we post a photo of most of the participants in the brainstorming meeting sponsored by the MacArthur Foundation at the Woods Hole Oceanographic Institution in July 2006 where the EOL concept took off. In the back row from the left: Brewster Kahle, John McCarter, Mark Costello, James Edwards, Jesse Ausubel; front row: John Hurley, David Patterson, Fred Grassle, Andrew Polaszek, Cristian Samper.
Encylopedia of Life opens, another step toward global commons of biodiversity knowledge
 On February 27, 2008, Encyclopedia of Life (EOL), “a web page for every species” officially launched, with over 30,000 species pages, mostly fish so far, and a diversity of links to internet resources including Biodiversity Heritage Library (2.9 million pages digitized). In case you missed it, there is a thrilling, award-winning video. Following Wikipedia model, EOL users are invited to become “curators” for one or more species pages, and later this year all are invited to submit content (photos, drawing, text, video, for example) for review. For an entertaining brief history of Wikipedia and why it keeps getting better see Nicholson Baker’s review of John Broughton’s Wikipedia: The Missing Manual in March 20, 2008 New York Review of Books.
On February 27, 2008, Encyclopedia of Life (EOL), “a web page for every species” officially launched, with over 30,000 species pages, mostly fish so far, and a diversity of links to internet resources including Biodiversity Heritage Library (2.9 million pages digitized). In case you missed it, there is a thrilling, award-winning video. Following Wikipedia model, EOL users are invited to become “curators” for one or more species pages, and later this year all are invited to submit content (photos, drawing, text, video, for example) for review. For an entertaining brief history of Wikipedia and why it keeps getting better see Nicholson Baker’s review of John Broughton’s Wikipedia: The Missing Manual in March 20, 2008 New York Review of Books.
 Most near-sun comets are now discovered by amateurs, using images downloaded from the Solar and Heliospheric Observatory (SOHO), a satellite launched December 2, 1995 as part of international collaboration between European Space Agency (ESA) and National Aeronautics and Space Administration (NASA). I expect that EOL and other open-access databases will lead to many more persons contributing to biodiversity science.
Most near-sun comets are now discovered by amateurs, using images downloaded from the Solar and Heliospheric Observatory (SOHO), a satellite launched December 2, 1995 as part of international collaboration between European Space Agency (ESA) and National Aeronautics and Space Administration (NASA). I expect that EOL and other open-access databases will lead to many more persons contributing to biodiversity science.
.
.
.
.
.
Solving puzzles of mitochondrial variation within and among species
What limits mitochondrial variation within species? In January 2008 PLoS Biology researchers from Karolinksa Institute, Sweden, and University of Newcastle upon Tyne, United Kingdom, report on an ingenious mouse model that shows strong purifying selection acting within a single generation, or even earlier, during embryogenesis. Stewart and colleagues employed “mtDNA mutator” mice which are homozygous defective for a nuclear gene which encodes a proof-reading subunit of mtDNA polymerase. These mice have increased levels of mtDNA mutations in all tissues, with mutations evenly distributed along all codon positions in mtDNA protein genes, accelerated senescence and “a number of phenotypes associated with mitochondrial diseases.” mtDNA mutator mice were backcrossed to wild-type mice to produce offspring that inherited defective mitochondria but whose nuclear genome is homozygous normal at the mtDNA polymerase locus. 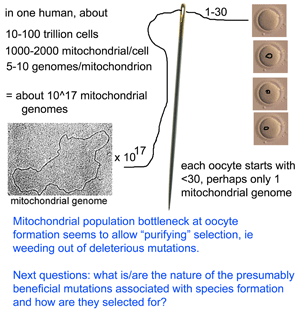 They then sequenced entire mitochondrial genomes from 190 of these progeny individuals in N2 to N6 generations (N2 is the first backcross that is homozygous normal at mtDNA mutator locus). To skip to the conclusion, most of the non-synonomous mitochondrial mutations were eliminated, leaving a pattern of excess synonymous mutations similar to that seen in human populations (which are the largest dataset so far for mitochondrial variation). The authors conclude that the mitochondrial population bottleneck known to occur at oogenesis, which deposits just one or few mitochondrial genomes per oocyte, means each mitochondrial genome must stand on its own so to speak, with the result that those eggs, embryos, or offspring harboring defective mitochondria will fail to survive. My figure at right tries to illustrate part of this process.
They then sequenced entire mitochondrial genomes from 190 of these progeny individuals in N2 to N6 generations (N2 is the first backcross that is homozygous normal at mtDNA mutator locus). To skip to the conclusion, most of the non-synonomous mitochondrial mutations were eliminated, leaving a pattern of excess synonymous mutations similar to that seen in human populations (which are the largest dataset so far for mitochondrial variation). The authors conclude that the mitochondrial population bottleneck known to occur at oogenesis, which deposits just one or few mitochondrial genomes per oocyte, means each mitochondrial genome must stand on its own so to speak, with the result that those eggs, embryos, or offspring harboring defective mitochondria will fail to survive. My figure at right tries to illustrate part of this process.
In the same issue, David Rand, Brown University, provides a lucid commentary on Stewart et al’s research putting it in the context of mitochondrial and evolutionary biology, and suggesting next steps. Among others, he notes “the new mouse study also begs new questions about positive selection on mtDNA. …it is interesting that no signature of a selective sweep leading to fixation of a novel mtDNA variant was evident in the data”.
Purifying selection against deleterious mutations enabled by an embryonic bottleneck may save mtDNA from “mutational meltdown”. Now we need to understand more about the positive selection on mtDNA that presumably occurs when species adapt to new environments or diverge. I believe that growing mtDNA databases in the form of COI barcodes from a diversity of organisms with varying size, lifespan, population size, and reproductive strategy, in a diversity of environments including marine, terrestrial, temperature, and tropical regions will help solve this puzzle.
The Encyclopedia of Life goes live
The Encyclopedia of Life, for which Jesse served as founding chairman, goes live! Reuters offers a good sample story about the debut of this promising macroscope. Please visit the EOL and provide feedback to the EOL about the site design and operation.
Testing DNA barcodes to help identify biodiversity hotspot plants including endangered and cryptic species
Plants challenge DNA barcoding. It has been difficult to identify candidate barcode regions that amplify readily and also distinguish among closely-related species. In 7 February 2008 PNAS (open access) researchers from University of Johannesburg; University of Costa Rica; Royal Botanic Gardens, Kew; and Imperial College, London, analyze potential barcode regions on specimens collected in plant biodiversity hotspots in Kruger National Park, South Africa, and Costa Rica. They initially tested eight candidate regions identified in earlier studies (coding regions accD, rpoC1, rpoB, ndhJ, ycf5, and matK, and non-coding trnH-psbA). Amplification was done according to earlier studies except that a different set of matK primers was used which appeared to be more effective. All eight regions were examined in 101 specimens representing 32 species of trees, shrubs, and achlorophyllous parasites from South Africa, and on 71 specimens representing 48 species of Costa Rican orchids (in all, 44 species with 2-7 specimens per species, and 36 species with one sample). Based on their analysis, the coding region matK with the new primer set and the non-coding region trnH-psbA were >90% effective in species identification. For reasons I do not understand, the authors favor unweighted pair group method with arithmetic mean (UPGMA) for analyzing genetic clustering, although they tested neighbor-joining, maximum likelihood, maximum parsimony, and Bayesian methods. Given the presumed advantages of a coding region barcode (ease of alignment, greater higher-level phylogenetic signal), Lahaye et al propose 5′ region of plastid gene matK as a first-pass standard barcode for plants. 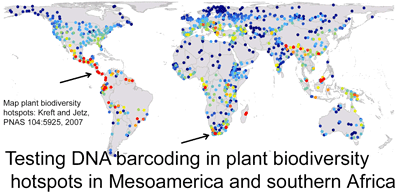
The authors then analyzed the 5′ matK barcode in a much larger sample of orchids: 1,566 specimens representing 1,084 Mesoamerican species. It is exciting that this is the largest test of candidate barcode variation within species for plants to date. They report 212 genetic clusters in UPGMA tree, of which “86 fully matched previously recognized species and a further 25 partially matched taxonomic species…an examination of these clusters reveals cryptic species, which need further taxonomic work”. I am unsure from this short report what “partially matched taxonomic species” are and how many possible cryptic species were identified. I look forward to a more detailed report on the DNA barcodes, morphology, and range distribution of this very large sample of Mesoamerican orchids. A DNA-based method for identifying non-flowering orchids and other plants could help protect many threatened species.
Science issue on cities
Our work in the 1980s on Cities and Their Vital Systems with Robert Herman, Cesare Marchetti, Alvin Weinberg, Brian Arthur, Nebojsa Nakicenovic and others seems to have acquired cult status, and caused Jesse’s inclusion in a video podcast (high and low bandwidth) to introduce the 8 February 2008 special issue of Science magazine dedicated to cities.The issue also includes an article by Paul Grant on Chauncey Starr’s concept of the supergrid for joint distribution of hydrogen and electricity.
New translation of classic Mendeleev article on origins of petroleum
Where did petroleum come from? How did it form? When? These are the first few questions the great scientist Dmitri Mendeleev asked in the chapter “On the origins of petroleum” in his book “Petroleum industry in Pennsylvania and Caucasus“. The year was 1877, 120 years after Mikhail Lomonosov pronounced that oil is a fossil fuel.
We are happy to post Veselin Kostov’s translation of this chapter together with a list of references on the abiogenic theory of petroleum origin (wherever our limited knowledge of Russian interrupted the continuous flow of Mendeleev’s thoughts and ideas, we’ve put XXX or ??). We leave to the reader to decide which theory holds more merit.