Jesse’s paper on “Renewable and Nuclear Heresies” appeared 25 July and attracted coverage in the UK Guardian newspaper.
News
Earth at Night
We post EARTH AT NIGHT a short note by Nadja Victor and Jesse Ausubel that has appeared in the debut issue of the Electronic Journal of Sustainable Development.
DNA-assisted discovery of new leopard in Borneo worries some taxonomists
Like a telescope that reveals hidden structures in the universe, genomic analysis is a window into biodiversity. For one, differences in DNA sequences help reveal how biodiversity is partitioned into the distinct populations we call species. In Frontiers Zool 29 May 2007, researchers from University of Wurzburg, US National Cancer Institute, and Arizona State University report on mitochondrial DNA and nuclear microsatellite differences between clouded leopards (Neofelis nebulosa) from Borneo (5 individuals), Sumatra (2 individuals), and mainland SE Asia (6 individuals). This report is a follow-up on two papers in December 2006 Current Biol which proposed separate species status for Bornean clouded leopards on the basis of differences in coat pattern and DNA. Wilting et al conclude their updated results “strongly support reclassification of clouded leopards into two distinct species N. nebulosa and N. diardi“. In addition to distinct coat patterns, the two lineages differ by 4.5% in mitochondrial coding genes (cytochrome b and ATPase-8), equivalent to or larger than genetic distances between the other well-recognized species of big cats in Panthera genus (lion, jaguar, tiger, leopard, snow leopard), suggesting the two lineages of clouded leopards have been separated for about 2.86 million years.
This sounds straightforward, but some taxonomists lament the increasing role of DNA in species discovery. In an editorial in current PLoS ONE, researchers from Imperial College insist the Bornean clouded leopard is not really new as it was “described by Cuvier in 1823.” Of course, by this criteria, most forms of larger animals will have been “described” by someone. Cuvier’s original work naming Felis diardi is three short paragraphs based on a single specimen and the illustration is unrecognizable.


To my reading, Meiri and Mace’s editorial implies that most of the important taxonomic work has already been done and if new genetic data appear to upset the traditional scheme, then it is being incorrectly interpreted. They note that there are another 144 mammal species shared between Borneo and the Malay Peninsula, thus “there could potentially be equivalent evidence to merit specific status for all of these; an outcome that would surely be unjustified”. An outcome that would surely be unjustified? This question needs to be answered by science, not by an appeal to taxonomic tradition. It may be that many island populations, which are now considered allopatric forms of widely distributed species, will turn out to be distinct species.
I close with the observation that just as genetic data can suggest splits it can also help reveal synonomies (multiple names that refer to the same species), suggest lumps, and identify forms that do NOT merit separate conservation status. For example, in Proc R Soc B 2005 Johnson et al apply mitochondrial DNA analysis to argue that the Cape Verde kite is not genetically distinct from the Black kite Milvus migrans and does not merit separate conservation status.
Mapping routes for DNA barcoding land plants
 Land plants challenge standardized DNA-based identification. Different groups of land plants are deeply divergent at the DNA level, yet there are relatively few sequence differences among closely-related species. Deep divergences make it difficult to design broad-range primers that amplify DNA from the many kinds of plants, and small differences among closely-related species mean longer sequences are needed to distinguish them. Plant mitochondrial genes including COI evolve too slowly to be useful. The best strategy appears likely to be a combination of 2 or 3 gene regions from the chloroplast genome. Chloroplasts are organelles which house the plants’ photosynthetic machinery and have their own genome, like mitochondria.
Land plants challenge standardized DNA-based identification. Different groups of land plants are deeply divergent at the DNA level, yet there are relatively few sequence differences among closely-related species. Deep divergences make it difficult to design broad-range primers that amplify DNA from the many kinds of plants, and small differences among closely-related species mean longer sequences are needed to distinguish them. Plant mitochondrial genes including COI evolve too slowly to be useful. The best strategy appears likely to be a combination of 2 or 3 gene regions from the chloroplast genome. Chloroplasts are organelles which house the plants’ photosynthetic machinery and have their own genome, like mitochondria.
In May 2007 Taxon, 19 researchers from 12 institutions in 7 countries (Brazil, Colombia, Denmark, Mexico, South Africa, U.K. and U.S.A.) report on tests of candidate barcode regions. Chase and co-investigators outline the rationale and results for selecting and testing potential land plant barcode regions. The finalists were winnowed down from more than 100 coding and non-coding regions in chloroplast DNA by testing 96 pairs of closely-related plant species to see which regions could be amplified and provide discrimination. Although the actual data are not shown in this short update, they summarize their results by proposing three chloroplast gene regions as a standard barcode for land plants: two coding regions, matK and rpoC1, and, either a third coding region, rpoB, or the non-coding psbA-trnH spacer region.
In June 2007 PLoS ONE, Kress and Erikcson, Smithsonian Institution, examine nine potential loci (8 plastid regions which includes the four final candidates in the Taxon paper, and nuclear gene ITS). In this analysis, as in Chase et al report, there are two steps: first, does the region amplify with a standard set of primers, and second, if so, does the sequence enable discrimination of closely-related species. In the 48 pairs of species examined, only two loci, trnH-psbA and rbcL-a exhibited more than 90% success with standard primers. Based on this admittedly small sample, the authors propose a “two-locus global DNA barcode for land plants” in which “rbcL-a provides a strong recognition anchor that will place an unidentified specimen into a family, genus, and sometimes species; the highly variable trnH-psbA spacer will futher narrow the corrrect species identification where rbcL-a lacks discrimination power.”
These are promising starts towards a standardized DNA barcode for land plants. More tests are needed, including analysis of variation within species, as both studies used single specimens for each target species.
Martha’s Vineyard Gazette 12 July 2007
The Martha’s Vineyard Gazette covered Jesse’s 12 July 2007 talk to the Vineyard Conservation Society about the Census of Marine Life.
Standardized mtDNA analysis to help identify exotic wildlife
 Many exotic pets are also endangered or threatened species. Global illegal trade in protected wildlife is estimated at $10 billion annually. The essential first step in international wildlife law enforcement is accurate species identification. In April 2007 Conservation Genetics researchers from Trent University, Ontario, and Toronto Zoo apply DNA barcoding to species identification of genus Brachypelma tarantulas from Mexico. “Brachypelma…tarantulas are popular pets as they are long-lived [15-25 years], brightly-colored, and tend to be docile…leading to over-harvesting from the wild.” Petersen and colleagues developed a method for recovering DNA from shed exoskeletons (exuviae), an improvement over the usual practice for DNA study of live tarantulas of “inducing limb autotomy” ie removing a leg! Short COI sequences (205 bp) from 23 individuals representing 8 of the 20 known Brachypelma species were analyzed. Even with this short sequence, all species formed well-supported nodes in a NJ tree (tree topology recovered with maximum parsimony and maximum likelihood methods was consistent with NJ results). The authors call for a “reference set of COI barcodes…fully vouchered and accessioned into a recognized collection”. They conclude that analyzing DNA from exuvia will enable field researchers to “sample individuals in situ without reducing the fitness of animals or reducing the population size” and aid in conservation of these iconic species and their habitats.
Many exotic pets are also endangered or threatened species. Global illegal trade in protected wildlife is estimated at $10 billion annually. The essential first step in international wildlife law enforcement is accurate species identification. In April 2007 Conservation Genetics researchers from Trent University, Ontario, and Toronto Zoo apply DNA barcoding to species identification of genus Brachypelma tarantulas from Mexico. “Brachypelma…tarantulas are popular pets as they are long-lived [15-25 years], brightly-colored, and tend to be docile…leading to over-harvesting from the wild.” Petersen and colleagues developed a method for recovering DNA from shed exoskeletons (exuviae), an improvement over the usual practice for DNA study of live tarantulas of “inducing limb autotomy” ie removing a leg! Short COI sequences (205 bp) from 23 individuals representing 8 of the 20 known Brachypelma species were analyzed. Even with this short sequence, all species formed well-supported nodes in a NJ tree (tree topology recovered with maximum parsimony and maximum likelihood methods was consistent with NJ results). The authors call for a “reference set of COI barcodes…fully vouchered and accessioned into a recognized collection”. They conclude that analyzing DNA from exuvia will enable field researchers to “sample individuals in situ without reducing the fitness of animals or reducing the population size” and aid in conservation of these iconic species and their habitats.
Bird in hand may need DNA reference library
 Even in well-studied groups such as birds, there are specimens that experts cannot identify. For example, the Pterodroma petrel shown at right was closely examined and photographed after it was captured on a cruise ship outside Maui in 2003, but not conclusively identified. More generally, some closely-related bird species cannot be reliably distinguished morphologically, particularly juveniles and adults in non-breeding plumage, limiting the value of banding efforts in determining population size, mortality, and range, for example.
Even in well-studied groups such as birds, there are specimens that experts cannot identify. For example, the Pterodroma petrel shown at right was closely examined and photographed after it was captured on a cruise ship outside Maui in 2003, but not conclusively identified. More generally, some closely-related bird species cannot be reliably distinguished morphologically, particularly juveniles and adults in non-breeding plumage, limiting the value of banding efforts in determining population size, mortality, and range, for example.
When morphologic identifications are routinely incomplete, it may be worthwhile to routinely analyze DNA. For birds this is usually simple as a single breast feather plucked from a live bird and stored dry at room temperature generally contains sufficient DNA for barcode analysis.
Of course, for reliable identifications, a comprehensive reference library of DNA sequences is essential. The need for a well-stocked library is highlighted by an article in April 2007 Ibis on skuas. Skuas are large, brown, gull-like predatory birds that nest in polar regions and migrate widely in open oceans, although their usual routes are not known. Young skuas do not reappear on breeding grounds until they are 3 years old, and any reports on non-breeding birds are of great interest. In an earlier paper (Ibis 146:95, 2004) using mitochondrial DNA analysis (698 bp of 12s and cytochrome b), the researchers concluded that 2 birds found in England in 2001 and 2002 constituted the first records of Brown Skuas (Stercorarius antarctica) in Europe. In this year’s follow-up paper, they dropped that conclusion, as sampling of a larger number of individuals showed that the 3 south polar skua species, S. antarctica, S. chilensis, and S. maccormicki are not distinguished by 12s/cytb mitochondrial DNA sequences.
 All told, this is a lot of academic effort for an identification question that could have been answered quickly by a web inquiry if there were a comprehensive library of avian DNA barcodes. So far, researchers have contributed about 8,600 DNA barcode sequences from about 1,800 of the 10,000 known avian species (see www.barcodingbirds.org ), helping create an enduring reference work that will be of long-term use by a large community of scientists, regulatory personnel, and general public who are interested in birds.
All told, this is a lot of academic effort for an identification question that could have been answered quickly by a web inquiry if there were a comprehensive library of avian DNA barcodes. So far, researchers have contributed about 8,600 DNA barcode sequences from about 1,800 of the 10,000 known avian species (see www.barcodingbirds.org ), helping create an enduring reference work that will be of long-term use by a large community of scientists, regulatory personnel, and general public who are interested in birds.
In closing, one interesting finding in the 2007 “retraction” paper is the general absence of mitochondrial genetic variation among south polar skua species. Two recent studies of southern polar skuas (Polar Biol 2006 29:153; J Ornithol 2006 147 (suppl):238 ) showed regular hybridization with normal reproductive success between C. antarctica and C. maccormicki, and analysis of mitochondrial hypervariable regions indicated “strong gene flow and little genetic differentiation among southern hemisphere taxa”, which is what population biologists usually say when they study single species. Biologists are generally loathe to “lump” species, but these findings suggest it may be more accurate to consider the south polar skuas as one species.
Helping the public by incorporating DNA barcodes into species descriptions
 A bewildering array of morphologic nuances are needed to identify species. DNA sequences, aka DNA barcodes, can also be identifiers, with the advantage that anyone with the right device can name a specimen without having to consult an expert or search through taxonomic keys which are generally indecipherable to the non-specialist. As DNA barcode identifications become more common, will this mean a loss of biologically important knowledge? To my reading, the answer is no, as the subtle differences in specialized structures used to identify organisms generally provide little insight into how organisms live and what they do. Thus it is exciting that standardized COI DNA barcodes are being incorporated into species descriptions as this will help democratize access to species names and the biological knowledge they represent.
A bewildering array of morphologic nuances are needed to identify species. DNA sequences, aka DNA barcodes, can also be identifiers, with the advantage that anyone with the right device can name a specimen without having to consult an expert or search through taxonomic keys which are generally indecipherable to the non-specialist. As DNA barcode identifications become more common, will this mean a loss of biologically important knowledge? To my reading, the answer is no, as the subtle differences in specialized structures used to identify organisms generally provide little insight into how organisms live and what they do. Thus it is exciting that standardized COI DNA barcodes are being incorporated into species descriptions as this will help democratize access to species names and the biological knowledge they represent.
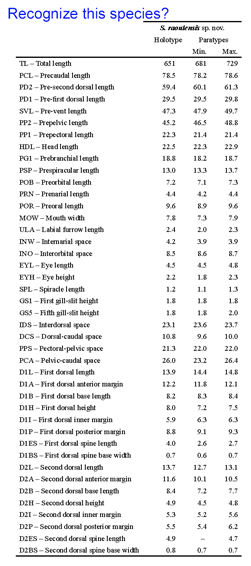 A 2007 monograph describes 11 new dogfish species in the genus Squalus from the Indo-West Pacific using “a rapid taxonomic approach” incorporating digital images, morphometric measurements, and DNA barcodes. Even for something as recognizable as a shark, it would be difficult for most persons to apply the morphometric measurements (example shown at left) used to characterize these new species. DNA barcoding has the potential to expand the pool of persons able to name shark species, helping those trying to understand shark biology and those assigned to enforce regulations that protect shark species. Biologists might choose to adopt DNA barcoding as a routine identification tool, rather than measuring “labial furrow length” or “internarial space”, for example.
A 2007 monograph describes 11 new dogfish species in the genus Squalus from the Indo-West Pacific using “a rapid taxonomic approach” incorporating digital images, morphometric measurements, and DNA barcodes. Even for something as recognizable as a shark, it would be difficult for most persons to apply the morphometric measurements (example shown at left) used to characterize these new species. DNA barcoding has the potential to expand the pool of persons able to name shark species, helping those trying to understand shark biology and those assigned to enforce regulations that protect shark species. Biologists might choose to adopt DNA barcoding as a routine identification tool, rather than measuring “labial furrow length” or “internarial space”, for example.
For many organisms, even those of economic importance, the number of persons who can apply the relevant morphologic tools is often very small, and the value of DNA barcoding as a widely-accessible tool potentially greater.
In Can Entomol 139:319 (2007) Jean-Francois Landry, Agriculture and Agri-Food, Canada, provides a taxonomic review of the leek moth genus Acrolepiopsis in North America which includes serious pests of onion and garlic crops.
The monograph includes detailed morphologic illustrations of male and female specimens, cocoons, pupae, and crop damage, and for the non-specialist, DNA barcodes of 30 individuals from 5 of the 6 species, and these are also publicly available on the BOLD website https://www.barcodinglife.org under “Published Projects” tab, including maps showing collection locations and photographs of the individual specimens.
As shown below in the dissections of male Acrolepiopsis genitalia, the morphologic illustrations can be beautiful, but the distinctive characters are not necessarily informative about the biology of the species.
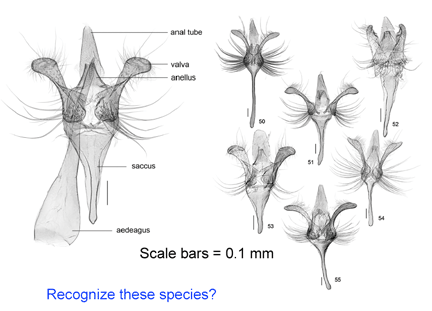
Iterative cycle of taxonomic and DNA sequence re-assessment essential for reliable barcode databases
 Like a map that is regularly updated, the reliability of DNA barcode databases will improve over time. To enable improvement, researchers have agreed to standardize on a particular region, to analyze multiple individuals from each species, and to revise DNA sequences and taxonomic labels as new information becomes available. By using specimens archived in museums, taxonomic identifications and DNA sequences can be re-checked. In March 2007 Med Vet Entomol 21:44, researchers from University of Wollongong, Australia, apply DNA barcoding to the identification of 9 species of forensically and medically important blowflies in family Calliphoridae. Calliphoridae blowflires cause disease in humans and domestic animals, and, in cases of murder or suspicious death, identification of blowfly species is a first step in determining the post-mortem interval. Identifications of adult flies requires specialized taxonomic knowledge and even experts have difficulty identifying egg and larval stages and the fragments of decomposed insects that may be all that is available in forensics. Nelson et al sequenced COI barcode region from legs of 52 adult flies representing 9 species in genus Chrysoma. The specimens were deposited in the Diptera collection at the School of Biological Sciences, University of Wollongong. NJ and Bayesisan analyses recovered each species as a distinct cluster, ie a well-supported reciprocally monophyletic group.
Like a map that is regularly updated, the reliability of DNA barcode databases will improve over time. To enable improvement, researchers have agreed to standardize on a particular region, to analyze multiple individuals from each species, and to revise DNA sequences and taxonomic labels as new information becomes available. By using specimens archived in museums, taxonomic identifications and DNA sequences can be re-checked. In March 2007 Med Vet Entomol 21:44, researchers from University of Wollongong, Australia, apply DNA barcoding to the identification of 9 species of forensically and medically important blowflies in family Calliphoridae. Calliphoridae blowflires cause disease in humans and domestic animals, and, in cases of murder or suspicious death, identification of blowfly species is a first step in determining the post-mortem interval. Identifications of adult flies requires specialized taxonomic knowledge and even experts have difficulty identifying egg and larval stages and the fragments of decomposed insects that may be all that is available in forensics. Nelson et al sequenced COI barcode region from legs of 52 adult flies representing 9 species in genus Chrysoma. The specimens were deposited in the Diptera collection at the School of Biological Sciences, University of Wollongong. NJ and Bayesisan analyses recovered each species as a distinct cluster, ie a well-supported reciprocally monophyletic group.
Early in the study, two complications were encountered. First, four specimens preliminarily identified as Ch. latifrons grouped with Ch. semimetallica. Second, a specimen identified as Ch. saffranea grouped with its closest relative Ch. megacephala. The adult voucher specimens were re-examined and the nuclear ITS gene was sequenced for these individuals. This confirmed that the first four specimens had been misidentified, in retrospect unsurprising given the close morphological similarity of the two species. The fifth anomalous individual was diagnosed as a hybrid based on comparison of nuclear and mitochondrial sequences. The authors conclude “the need for re-examination of misplaced specimens…highlights the importance of a voucher collection for all members of a barcode database.” I would add that the researchers’ willingness to re-examine taxonomic identifications and sequence data is just as important as the availability of voucher specimens.
Two other recent papers on blowfly identification with mitochondrial DNA showed incomplete resolution at species level, but in these the authors did not close the taxonomy-DNA circle, either by re-examining specimens or repeating sequence analysis. In Int J Legal Med 2007, Wells et al examined Lucilia sp blowflies, using published GenBank sequences and newly sequenced adult flies, and found overlap between all sister species of Lucilia for which 2 or more specimens were examined. It is unclear from this short note how many specimens were examined, their geographic origin, and whether they are stored as vouchers (online supplementary material is not available on publisher’s website at the time of this writing). There is no mention of re-examining their own specimens or analyzing other loci and of course it is not possible in most cases to confirm that taxonomic identifications and sequences in GenBank data are correct.
In July 2007 Proc R Soc B Whitworth et al examine 31 Protocalliphora individuals belonging to 12 species. Protocalliphora are Holoarctic species whose larva parasitize newly-hatched nestling birds. Blowfly larvae or pupae were collected from nests, and emergent flies were identified based on fly and pupal case morphology. As “the lower half of the abdomen of each fly was used for DNA sequencing” I assume this would not leave enough tissue for voucher specimens. They first attempted to construct a phylogeny using nuclear ITS, but found a very low level of substitutions between species and those found were all autoapomorphies, both of which suggest this is a very recently derived species complex. They were able to construct a phylogeny using amplified fragment length polymorphism (AFLP) mapping, with each species forming a reciprocally monophyletic group. This was then compared to mitochondrial sequence data.
Given that the title of the paper is “DNA barcoding cannot reliably identify….” it is inexplicable and also scientically inaccurate that they did NOT analyze the standard 648 bp COI barcode region, instead using a 374 bp fragment of COI and a 579 fragment of COII! It is likely that the results would be similar in any case, but their mitochondrial data cannot be combined with or directly compared to results with the growing DNA barcode libraries, which now contain about 260,000 barcode records from about 29,000 species. The mitochondrial sequences showed distinct clusters for 6 of the 12 species, and there were 2 other clusters comprising 2 and 4 species respectively. A separate analysis suggests these multi-species clusters reflect horizontal transfer of mitochondrial DNA among closely-related species as a result of Wolbachia infection, and the authors speculate that, since Wolbachia are found in “15-75% of insect species”, there may difficulty using DNA barcoding to resolve many insect species. To my reading, their data suggest this is a very recently derived species complex and hybridization among species is common. One of the utilities of DNA barcoding is to highlight exceptional groups, such as this one appears to be, deserving of further study. For the next studies on DNA barcoding in Lucilia and Protocalliphora, I hope the researchers retain voucher specimens and sequence the standard barcode fragment!
DNA barcodes suggest fractal nature of genome
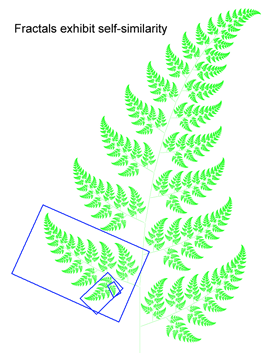 Growing data sets demonstrate DNA barcoding usually works, but why? Why does a very short stretch of DNA, such as a DNA barcode which usually represents less than one one-millionth of the genome, enable identification of most animal species? In computer language, Rod Page describes a DNA barcode as “embedded metadata“. Here I suggest an analogy to fractals, which might help convey what DNA barcodes reveal about how genomes are constructed.
Growing data sets demonstrate DNA barcoding usually works, but why? Why does a very short stretch of DNA, such as a DNA barcode which usually represents less than one one-millionth of the genome, enable identification of most animal species? In computer language, Rod Page describes a DNA barcode as “embedded metadata“. Here I suggest an analogy to fractals, which might help convey what DNA barcodes reveal about how genomes are constructed.
DNA barcoding usually works because patterns seen in very short DNA sequences usually reflect patterns seen in longer sequences. In this way, DNA barcodes demonstrate “self-similarity”, a fundamental property of fractals. In March 28, 2007 PLoS One, researchers from Concordia University, Quebec, analyze 849 complete animal mitochondrial genomes, comparing GC composition in 648 bp COI barcode region to GC composition in the mitochondrial genome as a whole. Min and Hickey found “such short sequences can yield important, and surprisingly accurate, information about the [mitochondrial] genome as a whole. In other words, for unsequenced genomes, 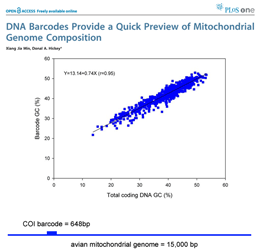 the DNA barcodes can provide a quick preview of the whole genome.” It will be of great interest to extend this analysis to compare mitochondrial barcodes to nuclear genomes; the general success of barcoding approach suggests there will be similarly close correlation.
the DNA barcodes can provide a quick preview of the whole genome.” It will be of great interest to extend this analysis to compare mitochondrial barcodes to nuclear genomes; the general success of barcoding approach suggests there will be similarly close correlation.
Overall, the patterning of barcode differences supports the emerging view that selective sweeps prune mitochondrial diversity within species and mitochondrial and nuclear co-evolution are tightly linked.
.
.
.
.