 What limits Japanese encephalitis virus (JEV) to its current range? JEV is a mosquito-transmitted flavivirus related to yellow fever and West Nile viruses that causes approximately 40,000 human cases annually in SE Asia. Although regular epidemics occur in islands off Papua New Guinea as close as 70 km to Australia and the major JEV vector in Papua New Guinea (PNG), Culex annulirostris, is found throughout Australia, there have only been sporadic cases in Australia and the disease has not become established there.
What limits Japanese encephalitis virus (JEV) to its current range? JEV is a mosquito-transmitted flavivirus related to yellow fever and West Nile viruses that causes approximately 40,000 human cases annually in SE Asia. Although regular epidemics occur in islands off Papua New Guinea as close as 70 km to Australia and the major JEV vector in Papua New Guinea (PNG), Culex annulirostris, is found throughout Australia, there have only been sporadic cases in Australia and the disease has not become established there.
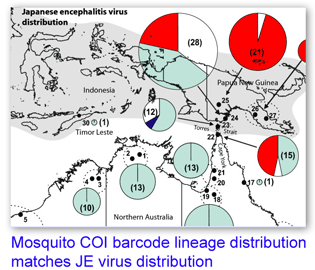 In 29 June 2007 BMC Evol Biol researchers analyzed mitochondrial COI and nuclear ITS 1 sequences in 273 mosquitos identified as Culex annulirostris or its close relatives Cx. palpalis and Cx. sitiens, collected at 30 locations in Australia and Papua New Guinea. Hemmerter et al found that 10% of morphological identifications were incorrect, based on ITS 1 sequences, and there was “100% agreement between the ITS 1 diagnostic and the COI sequence grouping of Culex spp.” Bayesian phylogenetic trees with COI showed “distinct geographically-structured lineages” (ie possible cryptic species) within the vector species Culex annulirostris, and two of the four Cx. annulirostris lineages are restricted to PNG, with a southern limit at the top of Australia’s Cape York peninsula, “which correlates exactly with the current southern limit of JEV activity”. Analysis of blood meals reveals the Australian Cx. annulirostris feed mainly on marsupials (PNG lineages feed on wild pigs which are the primary JEV reservoir), and laboratory studies indicate Australian Cx. annulirostris is an inefficient vector for JEV. As the authors note, it seems likely these genetically and biologically distinct lineages are likely different species.
In 29 June 2007 BMC Evol Biol researchers analyzed mitochondrial COI and nuclear ITS 1 sequences in 273 mosquitos identified as Culex annulirostris or its close relatives Cx. palpalis and Cx. sitiens, collected at 30 locations in Australia and Papua New Guinea. Hemmerter et al found that 10% of morphological identifications were incorrect, based on ITS 1 sequences, and there was “100% agreement between the ITS 1 diagnostic and the COI sequence grouping of Culex spp.” Bayesian phylogenetic trees with COI showed “distinct geographically-structured lineages” (ie possible cryptic species) within the vector species Culex annulirostris, and two of the four Cx. annulirostris lineages are restricted to PNG, with a southern limit at the top of Australia’s Cape York peninsula, “which correlates exactly with the current southern limit of JEV activity”. Analysis of blood meals reveals the Australian Cx. annulirostris feed mainly on marsupials (PNG lineages feed on wild pigs which are the primary JEV reservoir), and laboratory studies indicate Australian Cx. annulirostris is an inefficient vector for JEV. As the authors note, it seems likely these genetically and biologically distinct lineages are likely different species.
One limitation of this study is that the COI region analyzed does not match the COI barcode region. By my analysis the 538-bp fragment analyzed in this study starts at position 359 in COI. As the defined COI barcode region is 648 bp starting at position 58, there is only 289 bp overlap between the sequences in this study and COI barcodes. It appears generally straightforward to amplify COI barcodes from insects including mosquitos, so I hope the next study on genetic differences in human disease vectors will amplify the COI barcode region, as that will enable linking the results to the growing DNA barcode library, amplifying the power of the research itself.
I conclude that routine application of standardized genetic testing, ie DNA barcoding, will help in understanding the distribution of mosquito biodiversity, with implications for human health.

 In
In 
