Sponges are difficult to identify and classify. Many sponges “have a depauperate suite of morphologic characters and/or are plagued by morphological homoplasies” and vary according to environmental conditions, challenging identification at species level and stymying attempts to reconstruct evolutionary lineages. In Dec 2007 J Marine Biol Assoc UK researchers from Geoscience Centre Gottingen, Germany, and Queensland Museum, Australia, report on how DNA can help. Worheide and Erpenbeck describe the nascent Sponge Barcoding Project www.spongebarcoding.org, which aims to collect DNA signature sequences [COI barcodes] from all 8,000 known marine intertidal, deep sea, and freshwater sponge taxa. According to the authors “DNA barcoding will open up a new dimension and quality in biodiversity research and will become of vital importance for the survival and acknowledgement of sponge taxonomy and increase its reputation over the coming decades.” In addition to assisting species-level identifications, the authors posit the necessity of “DNA-assisted” taxonomy of sponges given the inability to construct convincing higher order classifications with morphologic characters.
Sponges are difficult to identify and classify. Many sponges “have a depauperate suite of morphologic characters and/or are plagued by morphological homoplasies” and vary according to environmental conditions, challenging identification at species level and stymying attempts to reconstruct evolutionary lineages. In Dec 2007 J Marine Biol Assoc UK researchers from Geoscience Centre Gottingen, Germany, and Queensland Museum, Australia, report on how DNA can help. Worheide and Erpenbeck describe the nascent Sponge Barcoding Project www.spongebarcoding.org, which aims to collect DNA signature sequences [COI barcodes] from all 8,000 known marine intertidal, deep sea, and freshwater sponge taxa. According to the authors “DNA barcoding will open up a new dimension and quality in biodiversity research and will become of vital importance for the survival and acknowledgement of sponge taxonomy and increase its reputation over the coming decades.” In addition to assisting species-level identifications, the authors posit the necessity of “DNA-assisted” taxonomy of sponges given the inability to construct convincing higher order classifications with morphologic characters.
An accompanying article analyzes COI barcode results for 166 specimens belonging to 65 species of Caribbean sponges. Similar to findings in other animal groups, the 584 bp COI fragment produced a gene tree similar to that with 28s rRNA, a slowly-evolving nuclear gene. In a ML analysis, some species had overlapping or shared sequences, which the authors point out may mean these are not “good species”, specimen identifications are incorrect, or that these species cannot be distinguished by a COI barcode alone. The sequences are published in GenBank and available individually through the Sponge Barcode website, and I hope the authors will also make their sequence and specimen data available on the Published Projects section of BOLD. This will allow access to the analytic and display software on the BOLD site, enable easy comparison of the sponge data set with that of other animals, and facilitate testing of other methods particularly for those species which are not distinguished in ML analysis.
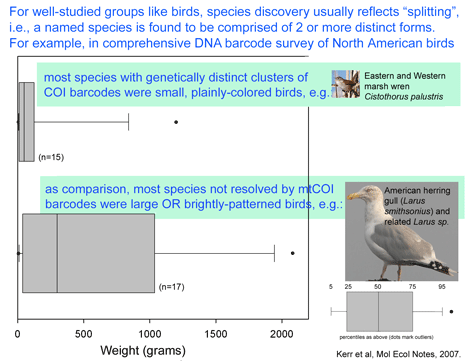
 Birds are relatively large, conspicuous, vocal, and mostly diurnal creatures, making it relatively easy for humans to tell apart. Even so, new species continue to be discovered; these usually represent distinct forms within what were thought to be single species. In
Birds are relatively large, conspicuous, vocal, and mostly diurnal creatures, making it relatively easy for humans to tell apart. Even so, new species continue to be discovered; these usually represent distinct forms within what were thought to be single species. In