The DNA barcode for animals is a 648 base pair (bp) fragment from the 5′ end of mitochondrial gene cytochrome c oxidase subunit I (COI). Does this relatively short mitochondrial sequence contain enough information to make evolutionary inferences about species limits, or is it a more of a rough survey method that needs to be confirmed by more data including from nuclear genes?
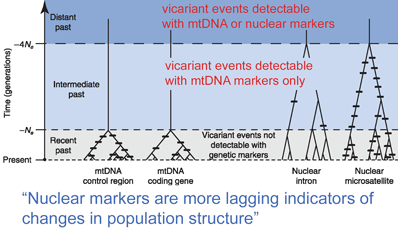 In April 2008 Mol Ecol researchers from University of Minnesota and American Museum of Natural History, New York, analyze utility of mitochondrial as compared to nuclear DNA for inferring recent evolutionary history. Zink and Barrowclough first apply population genetic theory and then look at real data from bird species.
In April 2008 Mol Ecol researchers from University of Minnesota and American Museum of Natural History, New York, analyze utility of mitochondrial as compared to nuclear DNA for inferring recent evolutionary history. Zink and Barrowclough first apply population genetic theory and then look at real data from bird species.
Based on mathematical population genetics, they find “mitochondrial loci are generally a more sensitive indicator of population structure than are nuclear loci,” primarily due to much smaller effective population size (Ne) for mitochondrial as compared to nuclear markers, which leads to more rapid sorting of differences among genetically isolated populations. Analysis of real-world data in 45 studies of differences among and within avian species confirms this expectation, ie either the patterning is consistent between mitochondrial and nuclear genes, or there are shallow mtDNA trees which are not yet reflected in nuclear genes. Reanalysis of one study, which appears to show a split in nuclear but not mitochondrial markers, suggests possible misinterpretation. Regarding other factors that could potentially lead to mistaken inferences about species limits based on mitochondrial DNA (NUMTs, sex-biased gene flow, introgression), experimental data suggests these are rarely important, at least in birds. The authors conclude “mtDNA patterns will prove to be robust indicators of population history and species limits.” Nuclear markers ARE important for deep gene trees, for detecting hybrids, and for “quantitative estimates…of rates of population growth and values of gene flow.”
Regarding length of mitochondrial sequence, this only has to be long enough to capture differences among closely-related species. Most populations that we recognize as species differ from their closest relatives by >1% in mitochondrial coding regions (corresponding to about 0.5 million years or more of reproductive isolation). At this level, even 100 bp is generally sufficient to distinguish most closely-related species, and a 648 bp COI barcode sequence should generally allow resolution of populations/species which have been reproductively isolated for much shorter periods of time.
 In
In  About 10% of named insect species are parasitoids, mostly wasps, but recognizing these often minute insects can be tricky. In
About 10% of named insect species are parasitoids, mostly wasps, but recognizing these often minute insects can be tricky. In  In
In  How many giraffes were onboard the Ark? Giraffes are classified as a single species, Giraffa camelopardalis, with five to nine subspecies proposed based on regional variation in pelage (coat pattern). In
How many giraffes were onboard the Ark? Giraffes are classified as a single species, Giraffa camelopardalis, with five to nine subspecies proposed based on regional variation in pelage (coat pattern). In 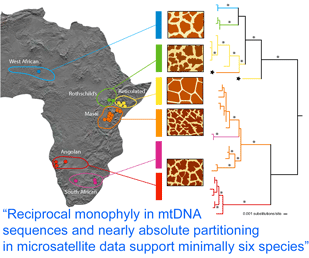 Analysis of 14 nuclear microsatellites from 381 individuals at 18 locations (it is not clear whether these are the same individuals as above) recovered the same six groups and suggested additional genetic subdivisions within some groups. Although at least some of the genetically and pelage-defined clusters have overlapping or adjacent ranges without geographic barriers, only three (0.8%) of individuals were identified as hybrids. These findings raise interesting questions about giraffe biology; for example, is there behavioral isolation perhaps based on visual recognition of pelage patterns?
Analysis of 14 nuclear microsatellites from 381 individuals at 18 locations (it is not clear whether these are the same individuals as above) recovered the same six groups and suggested additional genetic subdivisions within some groups. Although at least some of the genetically and pelage-defined clusters have overlapping or adjacent ranges without geographic barriers, only three (0.8%) of individuals were identified as hybrids. These findings raise interesting questions about giraffe biology; for example, is there behavioral isolation perhaps based on visual recognition of pelage patterns? 
 Just two years ago,
Just two years ago,