National Geographic’s weekly tv show Wild Chronicles featured the Census of Marine Life, including a few comments from Jesse.
Blog
We Welcome Veselin Kostov
We welcome our new Research Assistant Veselin Kostov!
Optimizing PCR primers for amphibian COI sequences
“Amphibians are globally in decline, yet there is still a tremendous amount of u nrecognized diversity” observed Vences et al in 2005 Phil Trans R Soc B 360:1859, the first report applying DNA barcoding to amphibian diversity. Vences and colleagues highlighted the pressing need for fast and reliable identification tools, including for eggs and larva, which are often unrecognizable morphologically.
nrecognized diversity” observed Vences et al in 2005 Phil Trans R Soc B 360:1859, the first report applying DNA barcoding to amphibian diversity. Vences and colleagues highlighted the pressing need for fast and reliable identification tools, including for eggs and larva, which are often unrecognizable morphologically.
Here I focus on one technical aspect of DNA barcoding amphibians, namely designing primers that amplify the target sequence from a broad range of species. Previous research had shown remarkable mitochondrial sequence diversity among closely-related amphibians, and even within what appear to be single species, some of which may represent cryptic species. In the 2005 Proc R Soc B paper, researchers used COI primers designed for invertebrates (Folmer et al 1994); suprisingly these “worked in a large proportion of specimens”. They concluded “We support attempts to build up a global and complete cox1 database of [animal] eukaryotes”.
In 2005 Frontiers Zool 2:5 the same group of researchers quantified their PCR amplification success on specimens from 38 individuals representing 20 amphibian species. Using a well-established primer set for vertebrate 16s (Palumbi et al 1991) 38 of 38 (100%) samples amplified; with 3 COI primer sets (1 for invertebrates, 2 for birds), 36 of 38 (95%) amplified, although there was only 50-70% success for the individual COI primer pairs. The authors did not attempt to design new primers for amphibians. They concluded “we strongly advocate use of 16s rRNA as standard DNA marker for vertebrates to complement COI”. This seems reasonable but the advantages of standardizing on a single gene call for an effort to design primers that amplify COI from amphibians before abandoning the field to 16s or some other marker.
In 2007 Mol Ecol Notes Smith and colleagues from University of Guelph analyzed 83 amphibian COI sequences in GenBank to design new primers. The 3′ ends of the forward and reverse primers bind at 1st or 2nd codon position G-C residues, which they found to be highly conserved among amphibian species, and each primer contains three 2-fold degenerate sites. Using this set, they amplified full-length PCR products from 267 of 377 specimens (71%) representing 39 amphibian species (including Triturus vulgaris illustrated at right), and recovered an additional 34 sequences (9%) using a “mini-barcode” primer set designed for butterflies. The authors comment “many of the specimens…which failed to amplify had been fixed in formalin or were collected more than 15 years ago”, so further work to test these primers on fresh material and a diversity of species is needed.
Amphibians are an exciting group. A comprehensive amphibian DNA barcode library will likely provide many, many new insights. I believe further work will help establish robust primer sets for amphibian COI sequences.
Future Knowledge of Life in Oceans
We post “Future Knowledge of Life in Oceans Past”, the published version of Jesse’s opening speech to the October 2005 Census of Marine Life conference on Oceans Past (see What’s New 25 October 2005). Congratulations to David Starkey, Poul Holm, and Michaela Barnard and the History of Marine Animal Populations community on completing the book (Oceans Past: Management Insights from the History of Marine Animal Populations, D. J. Starkey, P. Holm, and M. Barnard, eds., Earthscan, London and Sterling VA, 2008).
Telescopes and DNA Barcodes
Cold Spring Harbor Laboratory’s Banbury Conference Center hosted the pair of meetings in 2003 that gave birth to the DNA barcoding movement. Jesse opened the Banbury meeting 28 October 2007 on Using Barcode Data in Studies of Molecular and Evolutionary Dynamics with a talk on Telescopes, Microscopes, Macroscopes and DNA Barcodes.
New York Times “dot earth” Poppies
New York Times environment reporter Andrew Revkin has launched a new “dot earth” blot with an entry about population that draws on the work of the Program for the Human Environment about implosion and explosion published in the journal article “Human Population Dynamics Revisited with the Logistic Model: How Much Can Be Modeled and Predicted?”
Abstract
Decrease or growth of population comes from the interplay of death and birth (and locally, migration). We revive the logistic model, which was tested and found wanting in early-20th-century studies of aggregate human populations, and apply it instead to life expectancy (death) and fertility (birth), the key factors totaling population. For death, once an individual has legally entered society, the logistic portrays the situation crisply. Human life expectancy is reaching the culmination of a two-hundred year-process that forestalls death until about 80 for men and the mid-80’s for women. No breakthroughs in longevity are in sight unless genetic engineering comes to help. For birth, the logistic covers quantitatively its actual morphology. However, because we have not been able to model this essential parameter in a predictive way over long periods, we cannot say whether the future of human population is runaway growth or slow implosion. Thus, we revisit the logistic analysis of aggregate human numbers. From a niche point of view, resources are the limits to numbers, and access to resources depends on technologies. The logistic makes clear that for homo faber, the limits to numbers keep shifting. These moving edges may most confound forecasting the long-run size of humanity.
Non-invasive DNA recovery leaves tiny specimens intact
 Reference databases of DNA sequences used for species identification, ie DNA barcode libraries, are most powerful when the morphologic specimens are vouchered in a museum collection. This way, when there are puzzling results, DNA and morphologic specimens can be re-examined. However to date it has been challenging to recover DNA from small organisms without destroying them in the process.
Reference databases of DNA sequences used for species identification, ie DNA barcode libraries, are most powerful when the morphologic specimens are vouchered in a museum collection. This way, when there are puzzling results, DNA and morphologic specimens can be re-examined. However to date it has been challenging to recover DNA from small organisms without destroying them in the process.
In Mol Ecol Notes 9 aug 2007 researchers from US Department Agriculture and Smithsonian Institution, National Museum of Natural History, describe a uniform protocol for “nondestructive extraction of DNA from terrestrial arthropods” including ticks, spiders, beetles, flies, and bees. 1 to 4 h in a guanidium thiocyanate extraction buffer yielded amplifiable COI DNA from most specimens. Inspection of specimens after extraction including with phase contrast and scanning electron microscopy demonstrated preservation of most morphologic characters.
In Mol Ecol Notes 27 june 2007, UK researchers (University College, London, NERC Centre for Ecology and Hydrology, Oxford, and UK Environmental Agency) describe a rapid, non-destructive, chemical-free method for DNA recovery from blackflies, including adult, larval, and pupal forms. Hunter et al report brief (1 minute) sonication in sterile water yielded 66% success with COI barcode amplification and preserved morphologic details.
These reports are exciting in the methods they describe and in how they highlight the general value of extracting DNA and determining DNA barcode sequences as an integral part of preparing traditional morphologic vouchers.
Wood H to C ratio
The current madness for biofuels has raised again the question of the proper hydrogen-to-carbon ratio to use to index wood and other biomass against other fuels. We offer a note on the proper H:C ratio for wood.
Neotropical birds: Argentine researchers speed past halfway point
The Neotropics, comprising southern Mexico, Central America, Caribbean, and South America, is home to over 4,000 bird species, representing over 40% of world birds. In this post, Pablo Tubaro, Museo Argentino de Ciencias Naturales (MACN), Buenos Aires, Argentina, sends this update on DNA barcoding birds of Argentina:
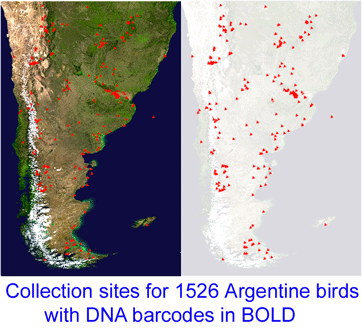 “This project, which started in December 2005, is a collaboration between MACN and the Biodiversity Insitute of Ontario/Canadian Center for DNA Barcoding (BIO/CCDB). In November 2006 the project was boosted by a grant from the Richard Lounsbery Foundation that supports expanded collecting efforts in Argentina, training of Argentine students at CCDB (2 trained so far), and establishment of a DNA laboratory at MACN.
“This project, which started in December 2005, is a collaboration between MACN and the Biodiversity Insitute of Ontario/Canadian Center for DNA Barcoding (BIO/CCDB). In November 2006 the project was boosted by a grant from the Richard Lounsbery Foundation that supports expanded collecting efforts in Argentina, training of Argentine students at CCDB (2 trained so far), and establishment of a DNA laboratory at MACN.
A special feature of this project is that it started literally from scratch. As there were no significant collections of frozen bird tissues with associated vouchers in Argentina, we started by resampling the country from north to south, conducting joint campaigns in collaboration with researchers from several North American institutions including American Museum of Natural History, Cornell University, Louisiana State University, Queen’s University, University of Alaska, and University of Kansas. At present our frozen tissue collection with associated vouchers includes more than 3100 samples and is growing rapidly. We will be doing field work at Iguazu National Park in November and December and aim to have collected 70% of Argentine birds by the year’s end.
Results so far show interspecific and intraspecific levels of divergence in COI squence are similar to published results with North American birds. In more than 98% of cases, the COI sequences belonging to different species do not overlap. In addition, in 3% of cases Argentine birds show distinct COI sequence clusters, suggesting the possible existence of cryptic species or geographical races that deserve species status. At this moment, four doctoral and post-doctoral fellowships have been requested or are already awarded by the National Research Council of Argentina (CONICET) and the National Science Foundation of Argentina (ANPCyT) to study in depth the phylogeographic structure of some of the interesting cases revealed by our DNA barcode survey.”
Congratulations to Pablo Tubaro and his team on their rapid progress in DNA barcoding Argentine birds, creation of a significant avian tissue and skin collection at MACN, and on recognition of the value of this work by science institutions in Argentina!
Lounsbery Political Attitudes
The Richard Lounsbery Foundation sponsored a new survey of political attitudes of American professors by Neil Gross (Harvard) and Solon Simmons (George Mason U.). Jesse offered opening remarks on behalf of Lounsbery at a lively symposium on “Professors and Their Politics” 6 October 2007 at Harvard to review and discuss the findings of the survey. Journalist Scott Jaschik provides and excellent account of the survey and symposium in Inside Higher Education.