By identifying species (leaves) and determining how they are related (branches), taxonomy aims to reconstruct the Tree of Life. To do so taxonomists must distinguish variation within species from that between species, and identify the shared characters that reflect evolutionary ancestry. These tasks require highly-specialized sets of knowledge and skill for each animal and plant group.
 One result is that it has been difficult to compare patterns of diversification between different branches in the Tree. One might ask, how finely and how evenly divided is biodiversity? Are the differences among and within mosquito species (3,400 species), for example, similar to the differences among and within fruit flies (6,200 species) or birds (10,000 species)? Broad application of DNA analysis is beginning to provide some insights. To enable these sorts of comparisons, a standardized locus is needed, as unique genes can solve local branching patterns, but do not allow easy comparisons between branches.
One result is that it has been difficult to compare patterns of diversification between different branches in the Tree. One might ask, how finely and how evenly divided is biodiversity? Are the differences among and within mosquito species (3,400 species), for example, similar to the differences among and within fruit flies (6,200 species) or birds (10,000 species)? Broad application of DNA analysis is beginning to provide some insights. To enable these sorts of comparisons, a standardized locus is needed, as unique genes can solve local branching patterns, but do not allow easy comparisons between branches.
Large-scale surveys of standardized genetic loci, including COI barcoding, commonly reveal distinct groups within what was thought to be a single species. There is also the converse finding that in some cases COI barcode sequences do not distinguish named species, but generally this strikes me as a relatively minor scientific problem that usually involves very closely-related species pairs and can be solved where needed with more DNA sequence, assuming the underlying taxonomy is correct and the named species really are distinct. The greater scientific challenge is finding multiple groups within what appear morphologically to be single species. In many cases so far, organisms with genetically distinct COI barcode clusters show associated biological differences signalling they represent different species.
In December 2007 Mol Ecol 16:4999, researchers from Museum of Comparative Zoology, Harvard, examine genetic differences in Aoraki denticulata, a tiny (2-3 mm) daddy longlegs or harvestman spider found in leaf litter widely through New Zealand. They determined COI barcode region sequences for 119 individuals from 17 localities in the mountainous northern part of South Island. The two described subspecies A. d. denticulata and A. d. major were genetically distinct. The surprising finding was there were at least 14 distinct clusters within A. d. denticulata, with a different group at almost every site, and 2 clusters at 3 of the sites. The differences between mtDNA clusters were as larger or larger than between other Aoraki species, up to 19.2%, but no morphologic differences were found even with electron microscopic scanning of males. Boyer et al observe “while it is conceivable that some of the geographically widespread populations…represent cryptic species, it is difficult to imagine that morphologically identical individuals from a single sample at a unique geographical point are not conspecific…it is hard to believe that almost every sampled locality would host at least one, if not two, cryptic species.”
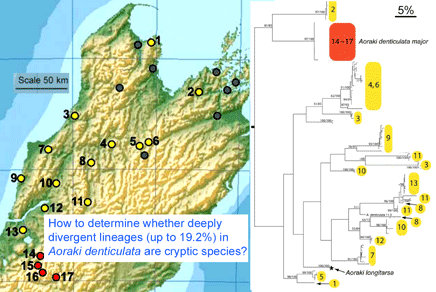 Continue reading “Standardized DNA analysis gives new vision of biodiversity, disturbing some”
Continue reading “Standardized DNA analysis gives new vision of biodiversity, disturbing some”
 mtDNA differences among these bivalves are remarkably large, even among species in the same genus. The differences among congeneric species in this sample (average 22%, range 12-42%) are larger than differences among entire class Aves (according to my analysis with BOLD software, COI differences among birds in different orders, such as penguins and hummingbirds for example, average 20%, with range 14-28%).
mtDNA differences among these bivalves are remarkably large, even among species in the same genus. The differences among congeneric species in this sample (average 22%, range 12-42%) are larger than differences among entire class Aves (according to my analysis with BOLD software, COI differences among birds in different orders, such as penguins and hummingbirds for example, average 20%, with range 14-28%). As of 28 january 2008, there are 341,825 barcode records from 35,798 species in the Barcode of Life Database (BOLD)
As of 28 january 2008, there are 341,825 barcode records from 35,798 species in the Barcode of Life Database (BOLD)  Lumpsuckers are globular, scaleless marine fish with bony tubercles on head and body, and a ventral sucking disc, derived from specialized pelvic fins, which allows them to adhere to environmental substrates. The genus Eumicrotremus comprises 16 species distributed in the Arctic and northern Atlantic and Pacific oceans; the commonest and most widespread in the north Atlantic is the Spiny lumpsucker E. spinosus, which was first described by Fabricius in 1776. A new subspecies E. s. eggvinii was described in 1956, based on a single specimen, and this was later elevated to species level “on the basis of wrinkled skin, numerous dermal warts and a large sucking disk, in addition to the low number of bony tubercles.”
Lumpsuckers are globular, scaleless marine fish with bony tubercles on head and body, and a ventral sucking disc, derived from specialized pelvic fins, which allows them to adhere to environmental substrates. The genus Eumicrotremus comprises 16 species distributed in the Arctic and northern Atlantic and Pacific oceans; the commonest and most widespread in the north Atlantic is the Spiny lumpsucker E. spinosus, which was first described by Fabricius in 1776. A new subspecies E. s. eggvinii was described in 1956, based on a single specimen, and this was later elevated to species level “on the basis of wrinkled skin, numerous dermal warts and a large sucking disk, in addition to the low number of bony tubercles.”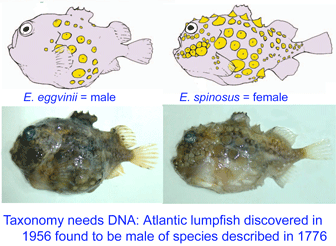 In August 2007
In August 2007