COI barcodes aim to enable identification of species, assigning unknown specimens to known species, and helping flag genetically divergent organisms that may represent new species. Might barcodes also help understand relationships among species?
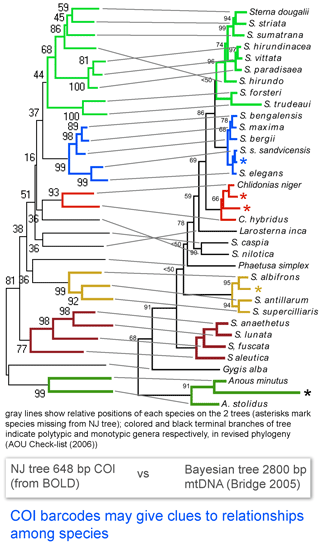 Here I look at one example from birds, comparing differences among COI barcodes to a recently revised phylogeny of terns (subfamily Sternini). According to American Ornithologists’ Union Check-list of North American Birds Supplement 47 (2006), “the data show that the genus Sterna as currently defined…is paraphyletic.”…”[W]e follow the recommendation of Bridge et al 2005 to resurrect four generic names currently placed in synonomy with Sterna.” The figure at left, taken from the 2005 paper by Bridge, Jones, and Baker, shows phylogenetic relationships based on 2800 bp of mtDNA from 33 species of terns (Bayesian tree with ML distances and ML boostrap support indices), and is juxtaposed to an NJ tree of COI barcodes from 29 of the same 33 taxa. The figure is colored according to the revised generic assignments (AOU 2006).
Here I look at one example from birds, comparing differences among COI barcodes to a recently revised phylogeny of terns (subfamily Sternini). According to American Ornithologists’ Union Check-list of North American Birds Supplement 47 (2006), “the data show that the genus Sterna as currently defined…is paraphyletic.”…”[W]e follow the recommendation of Bridge et al 2005 to resurrect four generic names currently placed in synonomy with Sterna.” The figure at left, taken from the 2005 paper by Bridge, Jones, and Baker, shows phylogenetic relationships based on 2800 bp of mtDNA from 33 species of terns (Bayesian tree with ML distances and ML boostrap support indices), and is juxtaposed to an NJ tree of COI barcodes from 29 of the same 33 taxa. The figure is colored according to the revised generic assignments (AOU 2006).
The topology of the COI NJ tree is similar to the larger data set tree, including that all currently recognized genera are reciprocally monophyletic, and most show similarly high boostrap values as in the Bayesian/ML analysis based on the larger data set.
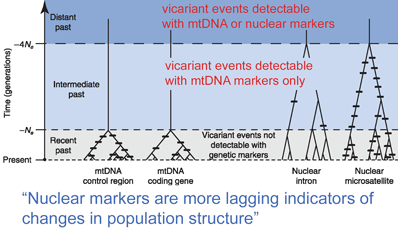
Of course mitochondrial DNA is widely used in analyzing relationships among animal species, including birds. Most of these studies are focused on relatively small groups of species, such as the tern study cited here. With growing DNA barcode libraries it will be increasingly possible to get at least a preliminary look at genetic relationships for large numbers of species (so far 2,393 avian species (24% of world birds) have barcode records in BOLD). This could be exciting!
 In
In  In
In  About 10% of named insect species are parasitoids, mostly wasps, but recognizing these often minute insects can be tricky. In
About 10% of named insect species are parasitoids, mostly wasps, but recognizing these often minute insects can be tricky. In  In
In  How many giraffes were onboard the Ark? Giraffes are classified as a single species, Giraffa camelopardalis, with five to nine subspecies proposed based on regional variation in pelage (coat pattern). In
How many giraffes were onboard the Ark? Giraffes are classified as a single species, Giraffa camelopardalis, with five to nine subspecies proposed based on regional variation in pelage (coat pattern). In 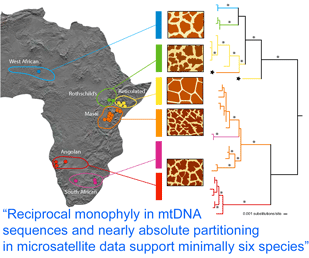 Analysis of 14 nuclear microsatellites from 381 individuals at 18 locations (it is not clear whether these are the same individuals as above) recovered the same six groups and suggested additional genetic subdivisions within some groups. Although at least some of the genetically and pelage-defined clusters have overlapping or adjacent ranges without geographic barriers, only three (0.8%) of individuals were identified as hybrids. These findings raise interesting questions about giraffe biology; for example, is there behavioral isolation perhaps based on visual recognition of pelage patterns?
Analysis of 14 nuclear microsatellites from 381 individuals at 18 locations (it is not clear whether these are the same individuals as above) recovered the same six groups and suggested additional genetic subdivisions within some groups. Although at least some of the genetically and pelage-defined clusters have overlapping or adjacent ranges without geographic barriers, only three (0.8%) of individuals were identified as hybrids. These findings raise interesting questions about giraffe biology; for example, is there behavioral isolation perhaps based on visual recognition of pelage patterns? 
 Just two years ago,
Just two years ago,