Next up, bees
Sunday, May 18th, 2008
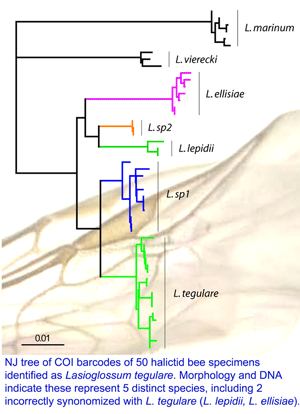 On 15 may 2008 an international assembly of bee experts gathered at York University and announced a new initiative to DNA barcode world’s bees. Some snippets from news reports:
On 15 may 2008 an international assembly of bee experts gathered at York University and announced a new initiative to DNA barcode world’s bees. Some snippets from news reports:
“According to York biology professor Laurence Packer, who’s leading the group’s efforts, precisely 19,231 different kinds of bees have been identified. But he thinks there might be another 5,000 or more species out there waiting.” (Toronto Star)
“There are two reasons why bee species would benefit from a barcode name tag. “Most of the specimens in museums are not identified, and the ones that are identified are only 60 to 70 percent correctly identified,” says Dr. Packer.” (University Affairs)
Early results, as in figure from poster at right, suggest that DNA barcoding will help sort out bee taxonomy and speed discovery of new species, with benefits to society and science. Bees are essential pollinators for many of the world’s plants, including many endangered species, and approximately 1/3 of world’s food is derived directly or indirectly from bee-pollinated plants.
In common with other Hymenoptera (bees, wasps, ants), bees exhibit haplodiploidy (males haploid, females diploid), many species are eusocial (live in large colonies with single queens), and have greatly accelerated rates of mitochondrial evolution. Are these factors causally linked? Looking at differences among and within world’s bees may help provide fundamental insight into mitochondrial biology and evolution.

 Authors Garros, Ngugi, Githeko, Tuno, and Yan collected anopheline larva near Kisumu in western Kenya, dissected stomach contents of third and fourth instar forms, extracted DNA, and amplified an 800 bp fragment of nuclear 18s rRNA. A separate PCR assay was used to confirm species identity (five were A. gambiae s.s. and 68 were sister species A. arabiensis). According to authors, 18s rRNA was analyzed rather than COI because “more sequences are available [for 18s than for COI] in databases for plants, fungi, and protists”. I note there are now many research groups working on “plants, fungi, and protists” so it should be possible to achieve greater resolution in this sort of study as the DNA barcode libraries are built up.
Authors Garros, Ngugi, Githeko, Tuno, and Yan collected anopheline larva near Kisumu in western Kenya, dissected stomach contents of third and fourth instar forms, extracted DNA, and amplified an 800 bp fragment of nuclear 18s rRNA. A separate PCR assay was used to confirm species identity (five were A. gambiae s.s. and 68 were sister species A. arabiensis). According to authors, 18s rRNA was analyzed rather than COI because “more sequences are available [for 18s than for COI] in databases for plants, fungi, and protists”. I note there are now many research groups working on “plants, fungi, and protists” so it should be possible to achieve greater resolution in this sort of study as the DNA barcode libraries are built up.
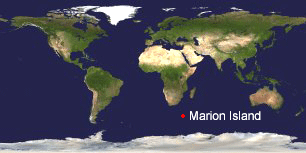 The challenge is to recognize invasive species before they become established. In
The challenge is to recognize invasive species before they become established. In  In April 2004, 3 noctuid moth larvae were found in an abandoned Wandering Albatross nest, a common habitat for one of the indigenous moth species. The larvae could be tentatively identified only to genus level and so rearing was attempted, with one larva dying after several months of pupating (as an aside, this is one example of how morphologic identifications can be laborious and/or incomplete, even for experts). The final larva was killed and preserved for DNA study; COI DNA barcode region was amplified using standard Folmer primers. The Marion Island moth larva barcode clustered with the 40 or so Black Cutworm Agrotis ipsilon sequences in BOLD, and was distinct from COI sequences of the other 18 Agrotis species in BOLD. Agrotis ipsilon is a common pest that feeds on a wide variety of plants. The authors conclude that Agrotis ipsilon is an established alien species with the potential to disrupt local ecosystems and that “steps be taken to eradicate the species from Marion Island.”
In April 2004, 3 noctuid moth larvae were found in an abandoned Wandering Albatross nest, a common habitat for one of the indigenous moth species. The larvae could be tentatively identified only to genus level and so rearing was attempted, with one larva dying after several months of pupating (as an aside, this is one example of how morphologic identifications can be laborious and/or incomplete, even for experts). The final larva was killed and preserved for DNA study; COI DNA barcode region was amplified using standard Folmer primers. The Marion Island moth larva barcode clustered with the 40 or so Black Cutworm Agrotis ipsilon sequences in BOLD, and was distinct from COI sequences of the other 18 Agrotis species in BOLD. Agrotis ipsilon is a common pest that feeds on a wide variety of plants. The authors conclude that Agrotis ipsilon is an established alien species with the potential to disrupt local ecosystems and that “steps be taken to eradicate the species from Marion Island.” Just posted, a
Just posted, a  Just as DNA analysis regularly overturns seemingly solid eyewitness identifications in crime investigations, routine DNA analysis can also help biologists avoid blunders. In
Just as DNA analysis regularly overturns seemingly solid eyewitness identifications in crime investigations, routine DNA analysis can also help biologists avoid blunders. In  On February 27, 2008,
On February 27, 2008, 
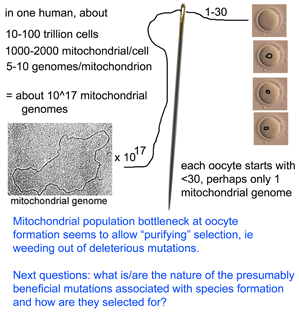 They then sequenced entire mitochondrial genomes from 190 of these progeny individuals in N2 to N6 generations (N2 is the first backcross that is homozygous normal at mtDNA mutator locus). To skip to the conclusion, most of the non-synonomous mitochondrial mutations were eliminated, leaving a pattern of excess synonymous mutations similar to that seen in human populations (which are the largest dataset so far for mitochondrial variation). The authors conclude that the mitochondrial population bottleneck known to occur at oogenesis, which deposits just one or few mitochondrial genomes per oocyte, means each mitochondrial genome must stand on its own so to speak, with the result that those eggs, embryos, or offspring harboring defective mitochondria will fail to survive. My figure at right tries to illustrate part of this process.
They then sequenced entire mitochondrial genomes from 190 of these progeny individuals in N2 to N6 generations (N2 is the first backcross that is homozygous normal at mtDNA mutator locus). To skip to the conclusion, most of the non-synonomous mitochondrial mutations were eliminated, leaving a pattern of excess synonymous mutations similar to that seen in human populations (which are the largest dataset so far for mitochondrial variation). The authors conclude that the mitochondrial population bottleneck known to occur at oogenesis, which deposits just one or few mitochondrial genomes per oocyte, means each mitochondrial genome must stand on its own so to speak, with the result that those eggs, embryos, or offspring harboring defective mitochondria will fail to survive. My figure at right tries to illustrate part of this process. 
 mtDNA differences among these bivalves are remarkably large, even among species in the same genus. The differences among congeneric species in this sample (average 22%, range 12-42%) are larger than differences among entire class Aves (according to my analysis with BOLD software, COI differences among birds in different orders, such as penguins and hummingbirds for example, average 20%, with range 14-28%).
mtDNA differences among these bivalves are remarkably large, even among species in the same genus. The differences among congeneric species in this sample (average 22%, range 12-42%) are larger than differences among entire class Aves (according to my analysis with BOLD software, COI differences among birds in different orders, such as penguins and hummingbirds for example, average 20%, with range 14-28%).