Early in Michael Crichton’s 1990 novel Jurassic Park, Dr. Henry Wu, chief scientist at Jurassic Park Research Insitute, showing visitors around his facility, displays “the actual structure of a small fragment of dinosaur DNA“. Astute readers pointed out Dr. Wu’s dinsosaur genetic resuscitation project was unlikely to succeed, as the sequence in Crichton’s novel was a fragment of the bacterial plasmid pBR322. They discovered this by feeding the “dinosaur sequence” into the online BLAST software engine, which searches the billions of base pairs of nucleotide sequences deposited in the amazing public resource of GenBank and the other international genetic databases, EMBL and DDBJ.
The power of genetic databases as identification tools rests on the quality of sequences and their annotations. Just as we need regularly updated maps for safe navigation, we need regularly updated genetic databases for accurate identifications.
One of the strengths of GenBank is that it serves as a permanent repository for genetic sequence data. As a result, GenBank is sometimes a permanent repository for faulty data. In a recent PLoS One paper, researchers from Goteborg University and Chalmers University of Technology, Sweden, and University of Tartu, Estonia, examined the taxonomic reliability of the 51,534 fungal internal transcribed spacer (ITS) sequences in the International Nucleotide Sequence Database (ie GenBank, EMBL, DDBJ). ITS is the most widely used locus for species identification in fungi.  The results show a “variegated picture of the taxonomic status of publicly indexed fungal sequences“. Taxonomic coverage is sparse: of the estimated 1.5 million fungi, less than 1% (9,684 species) are represented. Taxonomic data is lacking for many sequences (27% are not identified to species level), and most of the species-level identifications are unverifiable (82% are not linked to voucher specimens, 63% are not tagged with specimen country of origin, and 42% are marked as unpublished). Sequence comparisions suggest mislabeling is common (11% show best matches to congeneric but heterospecific sequences, and another 7% match among species of a different genus. Overall 10-21% of the INSD sequences have incorrect or unsatisfactory annotations.
The results show a “variegated picture of the taxonomic status of publicly indexed fungal sequences“. Taxonomic coverage is sparse: of the estimated 1.5 million fungi, less than 1% (9,684 species) are represented. Taxonomic data is lacking for many sequences (27% are not identified to species level), and most of the species-level identifications are unverifiable (82% are not linked to voucher specimens, 63% are not tagged with specimen country of origin, and 42% are marked as unpublished). Sequence comparisions suggest mislabeling is common (11% show best matches to congeneric but heterospecific sequences, and another 7% match among species of a different genus. Overall 10-21% of the INSD sequences have incorrect or unsatisfactory annotations.
It seems better to start over than to try to revise this Tower of Babel. Nilsson et al conclude “the large body of insufficiently identified fungi in INSD constitutes a silent plea for a wide and generalized sequencing effort of well-identified and -annotated [type] specimens residing in herbaria worldwide.” Toward this end, an All-Fungi Barcoding Initiative Workshop will be held 14-15 May 2007 at the Smithsonian Center for Research and Conservation, Fort Royal, Virginia. An international collection of researchers aim to hammer out how to build a reliable database, including which gene(s) should be adopted as standard barcode targets.
So far, DNA-based fungal identifications have primarily used ITS. Other nuclear genes have been used in some studies including the nuclear large ribosomal subunit, beta-tubulin, and elongation factor 1-alpha. It would be excellent if the fungal barcode database could link directly with those being built around the mitchondrial gene COI, which is effective for resolving most protozoan and metazoan (multicellular animal) species examined so far. In this regard it is exciting that a report by Seifert et al in 6 March 2007 Proc Natl Acad Sci USA shows COI provides species-level resolution similar to that for ITS, amplification was generally straightforward, and introns in the COI gene were found in only 2 of 370 Penicillium strains.
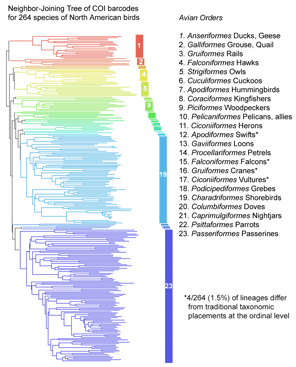 COI barcoding is a standardized approach to identifying species by DNA, helping resolve the “leaves” on the tree of life. Will the growing arrays of COI sequences also help provide insight into evolutionary history, the “branches” of the tree? I am struck that in some cases, simple genetic arithmetic with COI sequences creates trees very similar to modern phylogenies painstakingly created from multiple nuclear and mitochondrial genes, multiple morphologic characters, and exhaustive computerized analysis. Shown at right, a neighbor-joining analysis of
COI barcoding is a standardized approach to identifying species by DNA, helping resolve the “leaves” on the tree of life. Will the growing arrays of COI sequences also help provide insight into evolutionary history, the “branches” of the tree? I am struck that in some cases, simple genetic arithmetic with COI sequences creates trees very similar to modern phylogenies painstakingly created from multiple nuclear and mitochondrial genes, multiple morphologic characters, and exhaustive computerized analysis. Shown at right, a neighbor-joining analysis of 

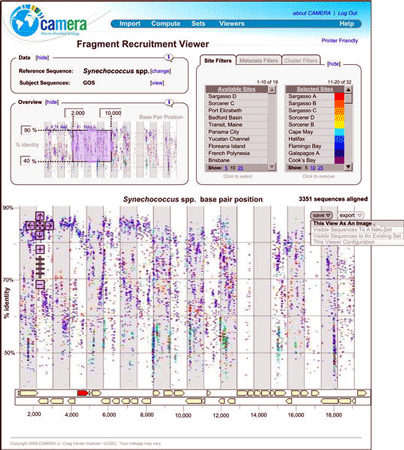


 A dozen articles in current issue of
A dozen articles in current issue of 
 The Indomalayan biogeographic region spans a vast area of tropical biodiversity and includes inumerable islands with high numbers of endemic species. A large scale genetic survey with DNA barcoding is likely to help lead to dramatic increases in species counts in particular and better understanding of biodiversity in general. Additional collecting may be particuarly important in this region, as it is at present the least well-represented in frozen tissue collections. There was strong enthusiasm among regional participants, and recognition the initiative has public appeal and the potential to engage new sources governmental support.
The Indomalayan biogeographic region spans a vast area of tropical biodiversity and includes inumerable islands with high numbers of endemic species. A large scale genetic survey with DNA barcoding is likely to help lead to dramatic increases in species counts in particular and better understanding of biodiversity in general. Additional collecting may be particuarly important in this region, as it is at present the least well-represented in frozen tissue collections. There was strong enthusiasm among regional participants, and recognition the initiative has public appeal and the potential to engage new sources governmental support. I look forward to organizational and scientific progress in this exciting region.
I look forward to organizational and scientific progress in this exciting region.  Two papers in early online
Two papers in early online 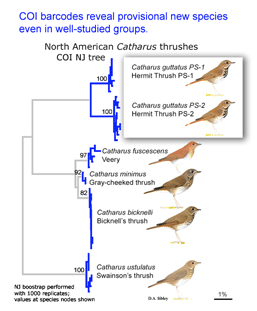 Birds being conspicuous, vocal, diurnal animals it is surprising that there are what appear to be overlooked species, even in an intensively-studied temperate region with relatively few species. Of course barcode clusters are not proof of species status, but to my knowledge all such divergent lineages either correspond to recognized species, or have subsequently been found to show biological covariants and have ultimately been granted species status.
Birds being conspicuous, vocal, diurnal animals it is surprising that there are what appear to be overlooked species, even in an intensively-studied temperate region with relatively few species. Of course barcode clusters are not proof of species status, but to my knowledge all such divergent lineages either correspond to recognized species, or have subsequently been found to show biological covariants and have ultimately been granted species status. I see the “barcode map of genetic diversity” as analogous to an astronomical sky map that uses just a slice of the electromagnetic spectrum. It does not contain all the information necessary to understand the universe, but by focusing on one part of the spectrum it enables results from various studies to be seamlessly combined and allows both large and small scale comparisions.
I see the “barcode map of genetic diversity” as analogous to an astronomical sky map that uses just a slice of the electromagnetic spectrum. It does not contain all the information necessary to understand the universe, but by focusing on one part of the spectrum it enables results from various studies to be seamlessly combined and allows both large and small scale comparisions. 

