Like a telescope that reveals hidden structures in the universe, genomic analysis is a window into biodiversity. For one, differences in DNA sequences help reveal how biodiversity is partitioned into the distinct populations we call species. In Frontiers Zool 29 May 2007, researchers from University of Wurzburg, US National Cancer Institute, and Arizona State University report on mitochondrial DNA and nuclear microsatellite differences between clouded leopards (Neofelis nebulosa) from Borneo (5 individuals), Sumatra (2 individuals), and mainland SE Asia (6 individuals). This report is a follow-up on two papers in December 2006 Current Biol which proposed separate species status for Bornean clouded leopards on the basis of differences in coat pattern and DNA. Wilting et al conclude their updated results “strongly support reclassification of clouded leopards into two distinct species N. nebulosa and N. diardi“. In addition to distinct coat patterns, the two lineages differ by 4.5% in mitochondrial coding genes (cytochrome b and ATPase-8), equivalent to or larger than genetic distances between the other well-recognized species of big cats in Panthera genus (lion, jaguar, tiger, leopard, snow leopard), suggesting the two lineages of clouded leopards have been separated for about 2.86 million years.
This sounds straightforward, but some taxonomists lament the increasing role of DNA in species discovery. In an editorial in current PLoS ONE, researchers from Imperial College insist the Bornean clouded leopard is not really new as it was “described by Cuvier in 1823.” Of course, by this criteria, most forms of larger animals will have been “described” by someone. Cuvier’s original work naming Felis diardi is three short paragraphs based on a single specimen and the illustration is unrecognizable.


To my reading, Meiri and Mace’s editorial implies that most of the important taxonomic work has already been done and if new genetic data appear to upset the traditional scheme, then it is being incorrectly interpreted. They note that there are another 144 mammal species shared between Borneo and the Malay Peninsula, thus “there could potentially be equivalent evidence to merit specific status for all of these; an outcome that would surely be unjustified”. An outcome that would surely be unjustified? This question needs to be answered by science, not by an appeal to taxonomic tradition. It may be that many island populations, which are now considered allopatric forms of widely distributed species, will turn out to be distinct species.
I close with the observation that just as genetic data can suggest splits it can also help reveal synonomies (multiple names that refer to the same species), suggest lumps, and identify forms that do NOT merit separate conservation status. For example, in Proc R Soc B 2005 Johnson et al apply mitochondrial DNA analysis to argue that the Cape Verde kite is not genetically distinct from the Black kite Milvus migrans and does not merit separate conservation status.




 A bewildering array of morphologic nuances are needed to identify species. DNA sequences, aka DNA barcodes, can also be identifiers, with the advantage that anyone with the right device can name a specimen without having to consult an expert or search through taxonomic keys which are generally indecipherable to the non-specialist. As DNA barcode identifications become more common, will this mean a loss of biologically important knowledge? To my reading, the answer is no, as the subtle differences in specialized structures used to identify organisms generally provide little insight into how organisms live and what they do. Thus it is exciting that standardized COI DNA barcodes are being incorporated into species descriptions as this will help democratize access to species names and the biological knowledge they represent.
A bewildering array of morphologic nuances are needed to identify species. DNA sequences, aka DNA barcodes, can also be identifiers, with the advantage that anyone with the right device can name a specimen without having to consult an expert or search through taxonomic keys which are generally indecipherable to the non-specialist. As DNA barcode identifications become more common, will this mean a loss of biologically important knowledge? To my reading, the answer is no, as the subtle differences in specialized structures used to identify organisms generally provide little insight into how organisms live and what they do. Thus it is exciting that standardized COI DNA barcodes are being incorporated into species descriptions as this will help democratize access to species names and the biological knowledge they represent. A
A 
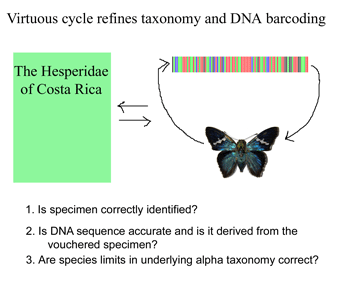 Like a map that is regularly updated, the reliability of DNA barcode databases will improve over time. To enable improvement, researchers have agreed to standardize on a particular region, to analyze multiple individuals from each species, and to revise DNA sequences and taxonomic labels as new information becomes available. By using specimens archived in museums, taxonomic identifications and DNA sequences can be re-checked. In
Like a map that is regularly updated, the reliability of DNA barcode databases will improve over time. To enable improvement, researchers have agreed to standardize on a particular region, to analyze multiple individuals from each species, and to revise DNA sequences and taxonomic labels as new information becomes available. By using specimens archived in museums, taxonomic identifications and DNA sequences can be re-checked. In  Growing data sets demonstrate DNA barcoding usually works, but why? Why does a very short stretch of DNA, such as a DNA barcode which usually represents less than one one-millionth of the genome, enable identification of most animal species? In computer language, Rod Page describes a DNA barcode as “
Growing data sets demonstrate DNA barcoding usually works, but why? Why does a very short stretch of DNA, such as a DNA barcode which usually represents less than one one-millionth of the genome, enable identification of most animal species? In computer language, Rod Page describes a DNA barcode as “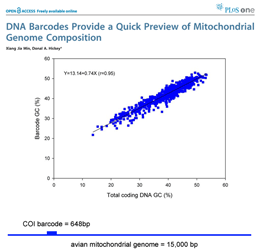 the DNA barcodes can provide a quick preview of the whole genome.” It will be of great interest to extend this analysis to compare mitochondrial barcodes to nuclear genomes; the general success of barcoding approach suggests there will be similarly close correlation.
the DNA barcodes can provide a quick preview of the whole genome.” It will be of great interest to extend this analysis to compare mitochondrial barcodes to nuclear genomes; the general success of barcoding approach suggests there will be similarly close correlation.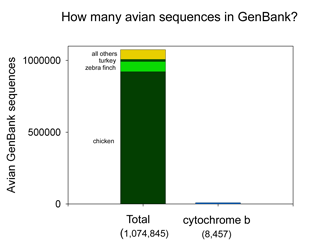 There are more than 1 million sequences in GenBank, but over 900,000 are from the Jungle Fowl (ie chicken, Gallus gallus), and another 85,000 from Zebra finch (Taeniopyga guttata) and Wild turkey (Meleagris gallopavo). That leaves about 67,000 sequences in total representing the rest of the approximately 10,000 species of world birds. According to Clements’ Birds of the World (including updates through 2006), there are 9,919 recognized species. The other world lists are very similar, and differ primarily in whether certain forms are recognized as species or subspecies and in assignment of generic names. I find it surprising there is not a single global taxonomic authority for bird species status, names, spelling, generic and family classification. As a comparison, medicine would be in great difficulty if there were not a single standard nomenclature for pathogenic bacteria.
There are more than 1 million sequences in GenBank, but over 900,000 are from the Jungle Fowl (ie chicken, Gallus gallus), and another 85,000 from Zebra finch (Taeniopyga guttata) and Wild turkey (Meleagris gallopavo). That leaves about 67,000 sequences in total representing the rest of the approximately 10,000 species of world birds. According to Clements’ Birds of the World (including updates through 2006), there are 9,919 recognized species. The other world lists are very similar, and differ primarily in whether certain forms are recognized as species or subspecies and in assignment of generic names. I find it surprising there is not a single global taxonomic authority for bird species status, names, spelling, generic and family classification. As a comparison, medicine would be in great difficulty if there were not a single standard nomenclature for pathogenic bacteria. 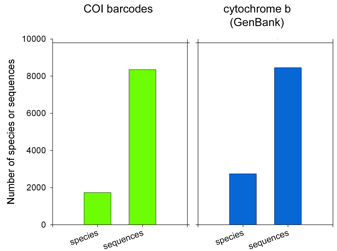 62,571 of the remaining 66,969 sequences are in the “CoreNucleotide” database (the others are unnamed genetic loci, either Expressed Sequence Tag (EST) or Genome Survey Sequence (GSS) records, and these will not be considered further here). Only 4,951 bird species are represented by any sequence (50% of world birds), and there are cytochrome b sequences for only 2,751 species (28% of world birds). Of species with cyt b sequences, 60% are represented by single sequences.
62,571 of the remaining 66,969 sequences are in the “CoreNucleotide” database (the others are unnamed genetic loci, either Expressed Sequence Tag (EST) or Genome Survey Sequence (GSS) records, and these will not be considered further here). Only 4,951 bird species are represented by any sequence (50% of world birds), and there are cytochrome b sequences for only 2,751 species (28% of world birds). Of species with cyt b sequences, 60% are represented by single sequences.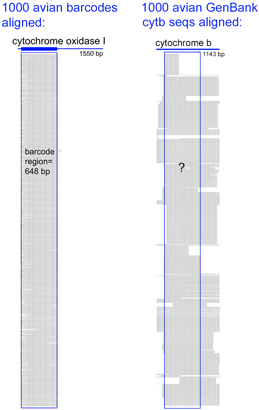 Virtues of the DNA barcode data set include that sequences are linked to vouchered museum specimens and their associated collecting data, sequence records include trace files to confirm sequencing accuracy, and most important all sequences can be directly compared because they derive from a standardized region. GenBank cyt b files include sequences of varying length and position along the gene. An alignment of 1000 avian COI barcodes and 1000 avian cyt b sequences hints at the power of a standardized approach.
Virtues of the DNA barcode data set include that sequences are linked to vouchered museum specimens and their associated collecting data, sequence records include trace files to confirm sequencing accuracy, and most important all sequences can be directly compared because they derive from a standardized region. GenBank cyt b files include sequences of varying length and position along the gene. An alignment of 1000 avian COI barcodes and 1000 avian cyt b sequences hints at the power of a standardized approach. A dream of many came to life this week with launch of
A dream of many came to life this week with launch of  The Scanning and Digitization Group will accelerate the work of the
The Scanning and Digitization Group will accelerate the work of the 
 In
In