“The first part of knowledge is getting the names right.” Chinese proverb quoted in Evolution of Insects, Grimaldi and Engel, 2005.
Species names are the primary entrance for accessing biological knowledge about organisms. However, the tangled bank of nomenclature created by 250 years of diverse communities of taxonomic specialists working largely in isolation challenges those seeking knowledge. It can be difficult to know what is already known. Identifying even well-studied organisms in backyards, such as North American ants for example, may require graduate-level training. As taxonomic knowledge moves increasingly onto the web, tools that enable non-specialists and specialists alike to access biological knowledge of organisms are beginning to be developed. In my view, the solution will be a combination of information science tools enabling access to biological literature together with a universal library of standardized genetic sequences, ie DNA barcodes, and simple technologies for barcode sequencing.
An exciting development in taxonomic information science is  uBio (Universal Biological Indexer and Organizer) www.ubio.org, “an initiative within the science library community to join international efforts to create and utilize a comprehensive and collaborative catalog of known names of all living (and once-living) organisms. The Taxonomic Name Server (10,699,999 NameBank records so far) catalogs names and classifications to enable tools that can help users find information on living things using any of the names that may be related to an organism.??”
uBio (Universal Biological Indexer and Organizer) www.ubio.org, “an initiative within the science library community to join international efforts to create and utilize a comprehensive and collaborative catalog of known names of all living (and once-living) organisms. The Taxonomic Name Server (10,699,999 NameBank records so far) catalogs names and classifications to enable tools that can help users find information on living things using any of the names that may be related to an organism.??”
The uBio site provides a sophisticated and enjoyable illustrated introduction (excerpt at right) to the variety of challenges in retrieving information using organism names. Another feature is Nomenclator Zoologicus, a searchable list of the names of genera and subgenera in zoology from the tenth edition of Linnaeus 1758 to the end of 2004, developed with Zoological Society of London. uBio is helping organize and index Encyclopedia of Life (“a web page for every species”) and Biodiversity Heritage Library (1.124 million pages digitized and on the web so far).
I close with an example from birds. Some taxonomic confusion reflects the struggle to integrate older works that use outdated taxon names or species limits with modern knowledge. Other discordances reflect lack of consensus among current experts. Given the intensity of scientific study and public interest in birds, it is surprising there is no single authoritative world checklist, especially since most of the differences at the species level reflect minor disagreements about generic assignment, a few cases of splitting/lumping, or differences in spelling. As one step until there is an expert consensus checklist, for those interested in birds, we have prepared an “ABBI Name Lookup” (Excel, 8 MB) file for harmonizing specimen lists that recognizes 2,462 synonyms, alternate and misspellings, and extinct species.

 Reference databases of DNA sequences used for species identification, ie DNA barcode libraries, are most powerful when the morphologic specimens are vouchered in a museum collection. This way, when there are puzzling results, DNA and morphologic specimens can be re-examined. However to date it has been challenging to recover DNA from small organisms without destroying them in the process.
Reference databases of DNA sequences used for species identification, ie DNA barcode libraries, are most powerful when the morphologic specimens are vouchered in a museum collection. This way, when there are puzzling results, DNA and morphologic specimens can be re-examined. However to date it has been challenging to recover DNA from small organisms without destroying them in the process. 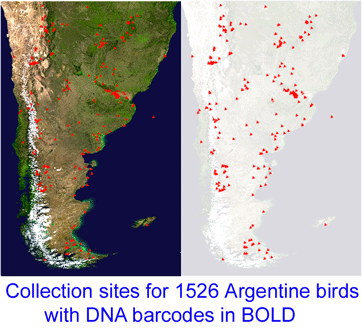 “This project, which started in December 2005, is a collaboration between MACN and the Biodiversity Insitute of Ontario/Canadian Center for DNA Barcoding (BIO/CCDB). In November 2006 the project was boosted by a grant from the Richard Lounsbery Foundation that supports expanded collecting efforts in Argentina, training of Argentine students at CCDB (2 trained so far), and establishment of a DNA laboratory at MACN.
“This project, which started in December 2005, is a collaboration between MACN and the Biodiversity Insitute of Ontario/Canadian Center for DNA Barcoding (BIO/CCDB). In November 2006 the project was boosted by a grant from the Richard Lounsbery Foundation that supports expanded collecting efforts in Argentina, training of Argentine students at CCDB (2 trained so far), and establishment of a DNA laboratory at MACN.  341 researchers from 44 countries gathered for the Second International Barcode of Life Conference, held at
341 researchers from 44 countries gathered for the Second International Barcode of Life Conference, held at 

 What limits Japanese encephalitis virus (JEV) to its current range? JEV is a mosquito-transmitted flavivirus related to yellow fever and West Nile viruses that causes approximately 40,000 human cases annually in SE Asia. Although regular epidemics occur in islands off Papua New Guinea as close as 70 km to Australia and the major JEV vector in Papua New Guinea (PNG), Culex annulirostris, is found throughout Australia, there have only been sporadic cases in Australia and the disease has not become established there.
What limits Japanese encephalitis virus (JEV) to its current range? JEV is a mosquito-transmitted flavivirus related to yellow fever and West Nile viruses that causes approximately 40,000 human cases annually in SE Asia. Although regular epidemics occur in islands off Papua New Guinea as close as 70 km to Australia and the major JEV vector in Papua New Guinea (PNG), Culex annulirostris, is found throughout Australia, there have only been sporadic cases in Australia and the disease has not become established there.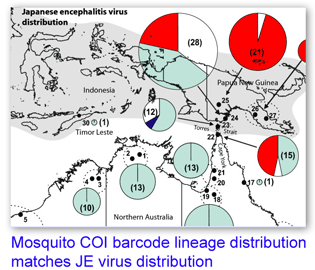 In
In 
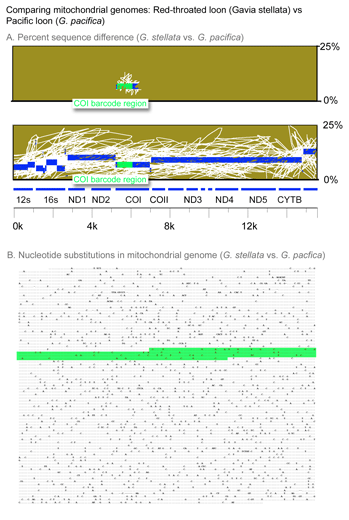
 In
In