 How many giraffes were onboard the Ark? Giraffes are classified as a single species, Giraffa camelopardalis, with five to nine subspecies proposed based on regional variation in pelage (coat pattern). In 21 dec 2007 BMC Biology (open access) researchers from University of California, Los Angeles; Center for Conservation Research, Omaha Zoo; and Mpala Research Centre, Kenya, investigate genetic variation in giraffes across African continent.
How many giraffes were onboard the Ark? Giraffes are classified as a single species, Giraffa camelopardalis, with five to nine subspecies proposed based on regional variation in pelage (coat pattern). In 21 dec 2007 BMC Biology (open access) researchers from University of California, Los Angeles; Center for Conservation Research, Omaha Zoo; and Mpala Research Centre, Kenya, investigate genetic variation in giraffes across African continent.
Using biopsy darts, the authors collected skin specimens from 266 giraffes at 19 localities in West, East, and South Africa. A 654 nucleotide region of mtDNA spanning cytb and control sequences was analyzed, revealing 35 haplotypes, and the remainder of the cytb gene (1709 bp total) was sequenced from one individual from each of the 35 haplotypes. The mtDNA sequences clustered into six reciprocally monophyletic lineages, which corresponded to groupings according to pelage pattern and regional location, and were largely concordant with subspecies designations. Genetic distances suggested these groups have been reproductively isolated for 0.3 to 1.6 MY, similar to calculated divergence times among other closely-related mammals.
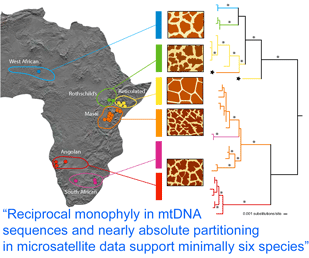 Analysis of 14 nuclear microsatellites from 381 individuals at 18 locations (it is not clear whether these are the same individuals as above) recovered the same six groups and suggested additional genetic subdivisions within some groups. Although at least some of the genetically and pelage-defined clusters have overlapping or adjacent ranges without geographic barriers, only three (0.8%) of individuals were identified as hybrids. These findings raise interesting questions about giraffe biology; for example, is there behavioral isolation perhaps based on visual recognition of pelage patterns?
Analysis of 14 nuclear microsatellites from 381 individuals at 18 locations (it is not clear whether these are the same individuals as above) recovered the same six groups and suggested additional genetic subdivisions within some groups. Although at least some of the genetically and pelage-defined clusters have overlapping or adjacent ranges without geographic barriers, only three (0.8%) of individuals were identified as hybrids. These findings raise interesting questions about giraffe biology; for example, is there behavioral isolation perhaps based on visual recognition of pelage patterns?
It is impressive that species can be overlooked in such large, boldly patterned, iconic animals. Might there be similar divisions within the numerous species of small, brown, rarely seen mammals? Routine DNA analysis of a standardized mtDNA region (aka DNA barcoding) will help discover how finely divided animal biodiversity is. Wilson and Reeder’s Mammal Species of the World, Third Edition lists 5,419 species, so this appears to be an achievable goal for our mammalian kin (list available online https://nmnhgoph.si.edu/msw/). I hope the authors include barcode region COI in their next analyses, so their data can be easily combined with other data sets, including the 28,560 mammalian barcode records in BOLD to date.

 Just two years ago,
Just two years ago,  In addition to helping identify what is already known, DNA analysis can reveal what would otherwise remain hidden. In
In addition to helping identify what is already known, DNA analysis can reveal what would otherwise remain hidden. In 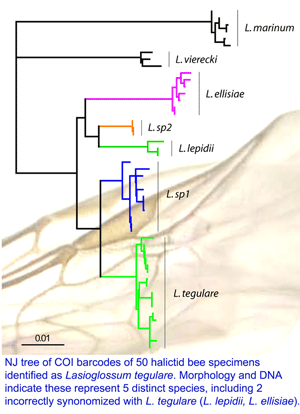 On 15 may 2008 an international assembly of bee experts gathered at York University and announced a new initiative to DNA barcode world’s bees. Some snippets from news reports:
On 15 may 2008 an international assembly of bee experts gathered at York University and announced a new initiative to DNA barcode world’s bees. Some snippets from news reports:
 Authors Garros, Ngugi, Githeko, Tuno, and Yan collected anopheline larva near Kisumu in western Kenya, dissected stomach contents of third and fourth instar forms, extracted DNA, and amplified an 800 bp fragment of nuclear 18s rRNA. A separate PCR assay was used to confirm species identity (five were A. gambiae s.s. and 68 were sister species A. arabiensis). According to authors, 18s rRNA was analyzed rather than COI because “more sequences are available [for 18s than for COI] in databases for plants, fungi, and protists”. I note there are now many research groups working on “plants, fungi, and protists” so it should be possible to achieve greater resolution in this sort of study as the DNA barcode libraries are built up.
Authors Garros, Ngugi, Githeko, Tuno, and Yan collected anopheline larva near Kisumu in western Kenya, dissected stomach contents of third and fourth instar forms, extracted DNA, and amplified an 800 bp fragment of nuclear 18s rRNA. A separate PCR assay was used to confirm species identity (five were A. gambiae s.s. and 68 were sister species A. arabiensis). According to authors, 18s rRNA was analyzed rather than COI because “more sequences are available [for 18s than for COI] in databases for plants, fungi, and protists”. I note there are now many research groups working on “plants, fungi, and protists” so it should be possible to achieve greater resolution in this sort of study as the DNA barcode libraries are built up.
 The challenge is to recognize invasive species before they become established. In
The challenge is to recognize invasive species before they become established. In  In April 2004, 3 noctuid moth larvae were found in an abandoned Wandering Albatross nest, a common habitat for one of the indigenous moth species. The larvae could be tentatively identified only to genus level and so rearing was attempted, with one larva dying after several months of pupating (as an aside, this is one example of how morphologic identifications can be laborious and/or incomplete, even for experts). The final larva was killed and preserved for DNA study; COI DNA barcode region was amplified using standard Folmer primers. The Marion Island moth larva barcode clustered with the 40 or so Black Cutworm Agrotis ipsilon sequences in BOLD, and was distinct from COI sequences of the other 18 Agrotis species in BOLD. Agrotis ipsilon is a common pest that feeds on a wide variety of plants. The authors conclude that Agrotis ipsilon is an established alien species with the potential to disrupt local ecosystems and that “steps be taken to eradicate the species from Marion Island.”
In April 2004, 3 noctuid moth larvae were found in an abandoned Wandering Albatross nest, a common habitat for one of the indigenous moth species. The larvae could be tentatively identified only to genus level and so rearing was attempted, with one larva dying after several months of pupating (as an aside, this is one example of how morphologic identifications can be laborious and/or incomplete, even for experts). The final larva was killed and preserved for DNA study; COI DNA barcode region was amplified using standard Folmer primers. The Marion Island moth larva barcode clustered with the 40 or so Black Cutworm Agrotis ipsilon sequences in BOLD, and was distinct from COI sequences of the other 18 Agrotis species in BOLD. Agrotis ipsilon is a common pest that feeds on a wide variety of plants. The authors conclude that Agrotis ipsilon is an established alien species with the potential to disrupt local ecosystems and that “steps be taken to eradicate the species from Marion Island.” Just posted, a
Just posted, a  Just as DNA analysis regularly overturns seemingly solid eyewitness identifications in crime investigations, routine DNA analysis can also help biologists avoid blunders. In
Just as DNA analysis regularly overturns seemingly solid eyewitness identifications in crime investigations, routine DNA analysis can also help biologists avoid blunders. In  On February 27, 2008,
On February 27, 2008, 