Analyzing shorter barcode sequences is an inexpensive way to link museum specimens with degraded DNA to the barcode database. In Molecular Ecology Notes July 2006, Hajibabaei et al first demonstrate in silico that COI sequences as short as 109 base pairs contain enough information to assign most specimens to known species, using simulated “minibarcodes” taken from two full-length barcode datasets. The researchers then analyzed the recovery and performance of various lengths of “minibarcodes” amplified from 33 dried and 91 ethanol-preserved insect specimens ranging in age from 1 to 21 years. As shown in the below, although full-length barcodes were recovered from only 24-39% of specimens, there was encouragingly high success amplifying shorter segments.
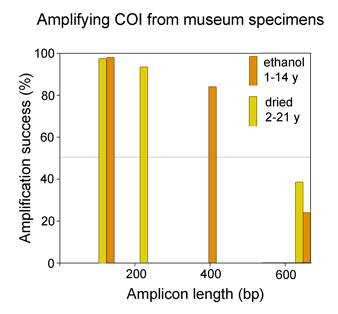
As expected from the in silico analysis, in most cases species could be distinguished as well as with full-length barcodes, ie sequences formed distinct non-overlapping clusters in a NJ tree. Hajibabaei et al’s results indicate that analyzing shorter minibarcode sequences can link museum specimens with degraded DNA to the gold standard full-length barcode database. Rather than spend time and money optimizing primers and amplification conditions on individual specimens, instead apply a general method that recovers a 100-400 bp fragment. They point out this approach will be useful “when barcoding reveals several cryptic species within what had been viewed as one species, and it is not morphologically evident which of them matches the holotype” and as “a cost-effective way of building barcode libraries with broad geographical coverage”. They caution that “very short barcode sequences are..valuable for the identification of old specimens from SELECTED NARROW taxonomic arrays” (emphasis added)
I agree a mini-barcode approach can be useful in certain situations, and emphasize their caution that it is not a substitute for a standardized full-length barcode database. First, if widely used, a minimalist approach could easily devolve into a Tower of Babel, with a hodgepodge of non-overlapping minibarcodes that cannot be compared to each other. Second, even if the minibarcodes were standardized so they all overlapped, a simple calculation implies that they would lump together most species with less than 1% sequence difference (in birds, this is about 15% of species). Less than 1% sequence difference means less than 6.5 diagnostic differences with a full-length barcode, and assuming randomly distributed substitutions, a shorter barcode could easily fail to capture any diagnostic differences.