Freshwater snails are intermediate hosts for schistosomiasis and flukes, trematode parasites that infect approximately 10% of world’s human population. Freshwater snails are also indicator species for water quality. Snail identification is essential for reducing disease burden and monitoring water quality.
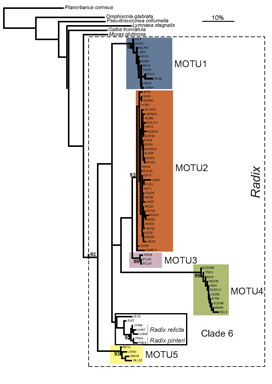 Researchers at the University of Frankfurt (November 2006 BMC Evol Biol 6:100) compared efficacy of morphologic and DNA-based taxonomy in freshwater snails in the genus Radix. Regarding Radix species in northwestern Europe, “species determination by shell morphology is difficult [and] unreliable…intraspecific variability of the putatively distinctive anatomical measurements largely overlaps among species” and identifications are “further complicated by recent nomenclatorial revisions”.
Researchers at the University of Frankfurt (November 2006 BMC Evol Biol 6:100) compared efficacy of morphologic and DNA-based taxonomy in freshwater snails in the genus Radix. Regarding Radix species in northwestern Europe, “species determination by shell morphology is difficult [and] unreliable…intraspecific variability of the putatively distinctive anatomical measurements largely overlaps among species” and identifications are “further complicated by recent nomenclatorial revisions”.
In their report, Pfenninger, Cordellier and Streit analyze morphology, mitochondrial COI and nuclear ITS-1 sequences, and describe breeding experiments with Radix snails collected at 60 sites throughout Europe. Using mtCOI sequences they found five MOTU (molecular operational taxonomic units), defined as “terminal clades with bootstrap support of 90% or more”. Populations of these MOTU overlapped broadly in geographic range and none corresponded to described species. Nuclear ITS sequences analyzed in a subset of specimens produced MOTU congruent with those generated by mtCOI.
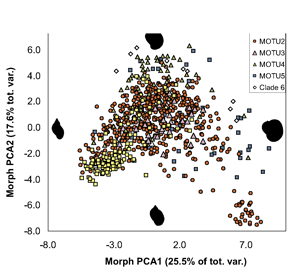 ALL crosses between individuals from the same MOTU population were viable, whereas NONE of crosses between individuals from different MOTU produced eggs. In morphometric analysis, Radix MOTU overlapped as shown at left, and in rearing experiments, shell shape changed in 4 of 5 populations, demonstrating phenotypic plasticity of putative morphologic characters. In northwestern European Radix snails, DNA trumps morphology.
ALL crosses between individuals from the same MOTU population were viable, whereas NONE of crosses between individuals from different MOTU produced eggs. In morphometric analysis, Radix MOTU overlapped as shown at left, and in rearing experiments, shell shape changed in 4 of 5 populations, demonstrating phenotypic plasticity of putative morphologic characters. In northwestern European Radix snails, DNA trumps morphology.
This work follows what might be a “best practices” pathway for single-locus mtDNA species discovery, aka DNA barcoding applied to species discovery:
1. COI sequence clusters (MOTU), found in analyzing multiple specimens from geographically widespread locations, are proposed as putative species.
2. COI clustering is congruent with nuclear sequence data.
3. COI clusters show corresponding biological differences, such as morphologic characters, behavioral differences, or breeding incompatibility.
In some cases a virus or bacteria is recognized to be the causative agent even though not all of Koch’s postulates have been fulfilled. In a similar way in some cases it might be desirable to recognize mtDNA clusters as representing species without fulfilling all of the above criteria.
 Here I offer one possible way of visualizing differences in barcode data sets using as an example the
Here I offer one possible way of visualizing differences in barcode data sets using as an example the 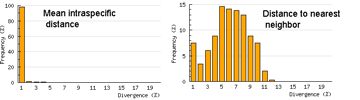 The histograms quickly show distances within most species are small and minimum distances between species are generally larger. Histograms are summaries with unlimited capacity. However, one might want to know more about individual species. For example, do species with higher intraspecific distances also show greater interspecific distances? One also wonders about the variation below 1% in both panels. In
The histograms quickly show distances within most species are small and minimum distances between species are generally larger. Histograms are summaries with unlimited capacity. However, one might want to know more about individual species. For example, do species with higher intraspecific distances also show greater interspecific distances? One also wonders about the variation below 1% in both panels. In 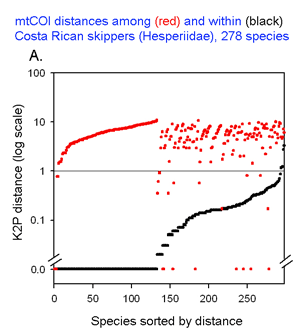
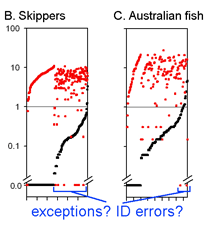 This graph is remarkably compressible, as shown by the small inset in the US county map above and in the figure at right. Here this is used to compare variation in Costa Rican skippers (278 species in 1 Family) to that in Australian fish (172 species in 1 Class) (
This graph is remarkably compressible, as shown by the small inset in the US county map above and in the figure at right. Here this is used to compare variation in Costa Rican skippers (278 species in 1 Family) to that in Australian fish (172 species in 1 Class) (


