Paul De Barro, CSIRO, Australia, recently posed the question “What is the expected level of mitochondrial variation within species?” The answer may be “almost none”. Results so far with DNA barcoding initiatives show average intraspecific variation in most animal species, whether saturniid moths or sand martins, is on the order of 0.5% or less. Here is my somewhat speculative set of inferences drawn from the finding of low variation within most animal species:
 1. Low intraspecific variation implies low effective population size (Ne); according to my back-of-the-envelope math, about 10,000 or so for most animal species. The apparent ceiling on Ne is low enough that census population size and species age, both of which might be expected to be determinants, do not contribute to intraspecific variation.
1. Low intraspecific variation implies low effective population size (Ne); according to my back-of-the-envelope math, about 10,000 or so for most animal species. The apparent ceiling on Ne is low enough that census population size and species age, both of which might be expected to be determinants, do not contribute to intraspecific variation.
2. What about species with larger average differences in mitochondrial DNA? Most are mosaics of reproductively-isolated or partially reproductively-isolated populations, some of which might be considered separate species. According to standard models of sequence evolution, it takes tens of thousands of years of reproductive isolation for distinct lineages of mitochondrial DNA to arise; significant morphological, ecological, and behavioral differences considered characteristic of separate species may arise over that length of time as well.
3. The paradoxical observation of large differences between species (indicating steady change) and small differences within (indicating change is constrained) implies that the pool of variants within a species changes steadily over evolutionary time scales. Like influenza virus, which regularly produces new variants that replace last year’s strains, the DNA sequences within breeding populations are continuously evolving, so that reproductive isolation over a sufficient period of time inevitably leads to genetic divergence. There may be morphologic stasis but there is no genetic stasis.
4. The usual absence of multiple lineages (with say >1% divergence in coding mtDNA) within breeding populations implies selection against hybrids and their offspring.
Now for some complex real data that challenge this simple model! In Proc R Soc B August 2007, researchers report on “Limited performance of DNA barcoding in a diverse community of tropical butterflies”. Elias and colleagues examined COI barcode region mtDNA sequences in 353 specimens from 57 species of ithomiine butterflies, most from 2 study sites in eastern Ecuador. Ithomiines are a tropical subfamily of approximately 360 species, virtually all of which are part of dizzyingly complex “rings” of Mullerian mimicry (all species distasteful) in which multiple species, some only distantly related, have nearly identical morpholgy. There is often marked geographic variation within what are considered single species such that different regional forms participate in different rings. For more appreciation, there are gorgeously illustrated research and other sites on ithomiines and other Mullerian mimics.
This exemplary study helps demonstrate the power of analyzing a standardized region, ie DNA barcoding, as their findings can be directly compared to results in other studies. In NJ analysis using the 273 study site specimens, the authors found that 44 of 57 (77%) of species formed well-supported (>50%) clusters. When sequences from non-local specimens were added to the analysis, and considering only species with more than one congener and with local and non-local sequences, 28 of 41 species (68%) formed distinct clusters. So one might mark down this group as challenging for DNA barcode approach to species identification.
One question is whether genetic diversity is more finely divided than current taxonomy recognizes. Differences within species sampled at distant geographic sites were as high as 8.5%, which the authors view as expected variation for tropical species with large census population sizes. Is this correct? Do larger populations support greater mitochondrial variation? According to report last year by Bazin et al Science 312:570 April 2006, the answer is no, but this conclusion seems not yet widely embraced. Following Bazin et al and the model outlined above, I suggest the genetically divergent forms reflect reproductively isolated allopatric populations and some might turn out to represent different species.
On the other end, some species had nearly identical COI sequences. Are these young species? The authors helpfully analyzed nuclear gene EF-1 alpha for most specimens and state that the nuclear gene sequence improved species-level identifications compared to mtCOI. On my inspection the published tree shows a similar overlap of EF-1 alpha gene sequences, which together with COI data suggests these are very closely-related young species. Recent work by some of the same authors Nature 441:868 14 June 2006 shows new species formation in just 3 generations in related Heliconidae butterflies through hybridization, so perhaps there are mechanisms that enable very rapid emergence of distinctive forms within these butterflies. There are presumably swarms of populations within many species that are distinctive in one form or another.
As this study shows, comparing relative and absolute differences in a standardized gene region is a useful approach for exploring the genetics of biodiversity. DNA barcode data sets can help address the question of whether population size influences mitochondrial sequence variation, and in turn the answer will help in understanding the patterning of genetic diversity among and within species. I look forward to more data on ithomiines and their relatives!
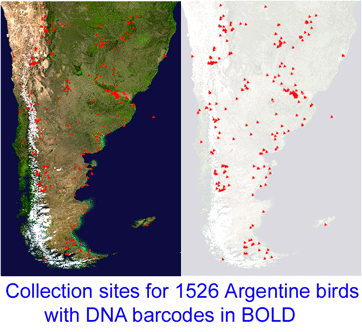 “This project, which started in December 2005, is a collaboration between MACN and the Biodiversity Insitute of Ontario/Canadian Center for DNA Barcoding (BIO/CCDB). In November 2006 the project was boosted by a grant from the Richard Lounsbery Foundation that supports expanded collecting efforts in Argentina, training of Argentine students at CCDB (2 trained so far), and establishment of a DNA laboratory at MACN.
“This project, which started in December 2005, is a collaboration between MACN and the Biodiversity Insitute of Ontario/Canadian Center for DNA Barcoding (BIO/CCDB). In November 2006 the project was boosted by a grant from the Richard Lounsbery Foundation that supports expanded collecting efforts in Argentina, training of Argentine students at CCDB (2 trained so far), and establishment of a DNA laboratory at MACN.  341 researchers from 44 countries gathered for the Second International Barcode of Life Conference, held at
341 researchers from 44 countries gathered for the Second International Barcode of Life Conference, held at 
 What limits Japanese encephalitis virus (JEV) to its current range? JEV is a mosquito-transmitted flavivirus related to yellow fever and West Nile viruses that causes approximately 40,000 human cases annually in SE Asia. Although regular epidemics occur in islands off Papua New Guinea as close as 70 km to Australia and the major JEV vector in Papua New Guinea (PNG), Culex annulirostris, is found throughout Australia, there have only been sporadic cases in Australia and the disease has not become established there.
What limits Japanese encephalitis virus (JEV) to its current range? JEV is a mosquito-transmitted flavivirus related to yellow fever and West Nile viruses that causes approximately 40,000 human cases annually in SE Asia. Although regular epidemics occur in islands off Papua New Guinea as close as 70 km to Australia and the major JEV vector in Papua New Guinea (PNG), Culex annulirostris, is found throughout Australia, there have only been sporadic cases in Australia and the disease has not become established there. In
In 
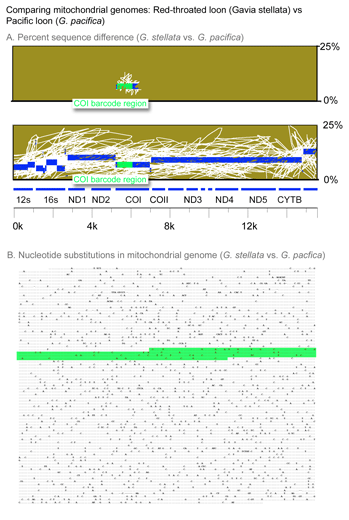
 In
In 


