In 9 September 2008 Proc Natl Acad Sci USA researchers from Brigham Young University and University of South Carolina report that nuclear pseudogenes, if not excluded from analysis, can confuse COI DNA barcoding studies. To my reading, this study re-iterates a well-understood hazard and proposes remedies that are already standard in most phylogenetic DNA work including DNA barcoding.
 Pseudogenes, first described by Jacq, Miller, and Brownlee in 1977, are non-functional genes that presumably arose from ancient duplication events and subsequent loss of function through accumulation of mutations. In sequencing studies, pseudogenes of protein coding genes are usually easily distinguished from their functional counterparts as they harbor insertions, deletions, and/or point mutations that interrupt the reading frame.
Pseudogenes, first described by Jacq, Miller, and Brownlee in 1977, are non-functional genes that presumably arose from ancient duplication events and subsequent loss of function through accumulation of mutations. In sequencing studies, pseudogenes of protein coding genes are usually easily distinguished from their functional counterparts as they harbor insertions, deletions, and/or point mutations that interrupt the reading frame.
Pseudogenes derived from mitochondrial DNA, often called numts (nuclear copies of mtDNA) were first reported by Gellissen et al in 1983. A search of NCBI PubMed for “mitochondrial pseudogenes” shows 282 articles and 12 review articles over the past 25 years.
Song and colleagues analyzed mitochondrial COI sequences in grasshoppers (single individuals of four species representing different Acrididae subfamilies) and cave crayfish (119 individuals of four species in genus Orconectes collected at 56 localities in southeastern US). Most of the analyses involved sequencing of cloned PCR products, which adds a level of complexity and is unlike any DNA barcoding study I am aware of. To skip to the conclusion, the authors emphasize that if numts generated by PCR amplification of mtCOI are NOT excluded, then it will confuse DNA barcoding or other phylogenetic studies. Since most of the numts generated in this study were easily recognized I do not understand why they did so much work (in all they sequenced 125 grasshopper clones and 560 crayfish clones) to reach this sensible but obvious conclusion.
First, grasshoppers. The authors amplified a subsegment of the COI barcode region (439 vs 648 bp in full-length barcode region; shorter amplicons are more likely to represent pseudogenes). The amplified products from the four individual grasshoppers were cloned, and 30 clones/species were sequenced, generating an average of 15 unique haplotypes per species. Of these, 97.3% had stop codons, meaning they could be immediately excluded as not representing true mtCOI sequences. A full-length barcode sequence was amplified from 1 species, and cloned products yielded 19 paralogues (ie obvious pseudogenes).
Second, crayfish. The researchers amplified the full-length COI barcode region from 172 individuals using Folmer primers. “For 93 individuals, we were able to obtain clean COI sequences; however, 79 individuals from southern populations of O. australis and O. barri yielded ambiguous sequences.” To my reading, the next step would be to stop there and find different primers or PCR conditions that did not generate ambiguous sequences (indicating that more than one COI-like template was being amplified). Instead the authors proceeded to clone products from individuals that yielded ambiguous results and also from those with clean sequences “to determine whether numts were present but not being detected without cloning.” Not surprisingly, they found probable numts in all 4 species of crayfish, and interestingly some of the clones did NOT contain stop codons (ie might be mistaken for functional COI sequences). These apparent numts, which might be easily overlooked, came from the 2 species with ambiguous results on sequencing of uncloned products, which I take as further evidence that it would have been better to develop a different COI amplification protocol, assuming the goal is to accurately determine the barcode sequence.
 Among other quality control standards in Barcode of Life Database (BOLD), COI sequences with stop codons, such as found in most pseudogenes in this study, are automatically flagged, signalling the researcher to re-check the data.
Among other quality control standards in Barcode of Life Database (BOLD), COI sequences with stop codons, such as found in most pseudogenes in this study, are automatically flagged, signalling the researcher to re-check the data.
Finally, it may be that some of what the authors call numts instead reflect heteroplasmy, ie differences among individual mitochondrial DNAs. Like static noise generated when you turn the volume up all the way, cloning is likely to reveal various mutations in some of the 10^17 or so mitochondrial genomes present in eukaryotic organisms. Looking ahead, it seems to me that the authors have missed an opportunity to contribute protocols or sequences that could be applied by other researchers to DNA barcoding of grasshoppers or crayfish.
 In October 2008 Wired reporter Gary Wolf profiles birth and rapid growth of standardized DNA-based species identification (ie DNA barcoding). His article centers around time spent in Costa Rica with Dan Janzen, Winnie Hallwachs, and their band of parataxonomists in Area de Conservacion Guanacaste; additional legwork includes visits to worried taxonomists at University of California Berkeley (“Honestly, I never thought it would get this far,” says Kipling Will), and University of Guelph, Ontario. He concludes with an evocative analogy: “barcodes are not just devices to put names on animals; they are also clever traps to catch all the people in the world whose curiosity impels them toward data as if toward light.”
In October 2008 Wired reporter Gary Wolf profiles birth and rapid growth of standardized DNA-based species identification (ie DNA barcoding). His article centers around time spent in Costa Rica with Dan Janzen, Winnie Hallwachs, and their band of parataxonomists in Area de Conservacion Guanacaste; additional legwork includes visits to worried taxonomists at University of California Berkeley (“Honestly, I never thought it would get this far,” says Kipling Will), and University of Guelph, Ontario. He concludes with an evocative analogy: “barcodes are not just devices to put names on animals; they are also clever traps to catch all the people in the world whose curiosity impels them toward data as if toward light.” An article in October 2008 Scientific American, with Sci Am’s trademark excellent illustrations, (web version; pdf) examines hows and whys of DNA-based future of species identification (I am co-author with Paul Hebert). After discussing the many practical applications for identifying known species, we conclude with our own analogy: “Just as the speed and economy of aerial photography caused it to supplant ground surveys as the first line of land analysis, DNA barcoding can be a rapid, relatively inexpensive first step in species discovery.”
An article in October 2008 Scientific American, with Sci Am’s trademark excellent illustrations, (web version; pdf) examines hows and whys of DNA-based future of species identification (I am co-author with Paul Hebert). After discussing the many practical applications for identifying known species, we conclude with our own analogy: “Just as the speed and economy of aerial photography caused it to supplant ground surveys as the first line of land analysis, DNA barcoding can be a rapid, relatively inexpensive first step in species discovery.” In
In  As a test of how DNA barcoding might work for the interested ant novice, I collected the tiny specimen at left in Rincon, Puerto Rico, and submitted its COI barcode to
As a test of how DNA barcoding might work for the interested ant novice, I collected the tiny specimen at left in Rincon, Puerto Rico, and submitted its COI barcode to  In
In 
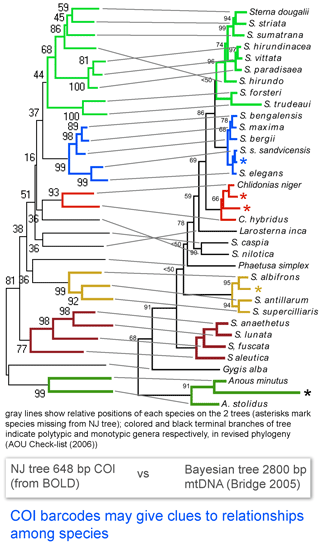 Here I look at one example from birds, comparing differences among COI barcodes to a recently revised phylogeny of terns (subfamily Sternini). According to
Here I look at one example from birds, comparing differences among COI barcodes to a recently revised phylogeny of terns (subfamily Sternini). According to 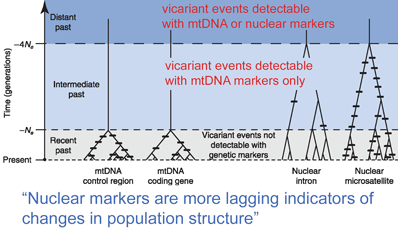 In
In  In
In  About 10% of named insect species are parasitoids, mostly wasps, but recognizing these often minute insects can be tricky. In
About 10% of named insect species are parasitoids, mostly wasps, but recognizing these often minute insects can be tricky. In  In
In